Синтез непредельных карбонильных соединений
Оглавление
Введение
1. Реакции с участием енолов
и енолят-ионов
1.1 Нуклеофильное замещение с участием енолят-анионов
1.2 Конденсации енолят-анионов
1.2.1 Альдольная конденсация
1.2.2 Внутримолекулярная циклизация дикетонов
1.2.3 Перекрестная альдольная конденсация
1.2.4 Реакция Кляйзена-Шмидта
2. Электрофильное присоединение
2.1 Гидратация Винилацетилена
3. Нуклеофильное
присоединение
3.1 Реакции α,β-непредельных карбонильных
соединений с литийалкилкупратами
4. Реакции отщепления
4.1 Дегидратация спиртов
4.1.1 Механизмы дегидратации спиртов
4.1.2 Дегидратация глицерина в присутствии
гидросульфата калия
5. Реакции окисления
5.1 Окисление первичных и вторичных спиртов
5.1.1 Окисление первичных и вторичных спиртов с
помощью ДМСО
5.1.2 Окисление вторичных спиртов по Оппенауэру
5.2 Окисление одноатомных непредельных спиртов
(алкенолы и алкинолы)
6. Свободнорадикальное
замещение
6.1 Каталитическое окисление алкенов
7. Перициклические реакции
7.1 Диеновый синтез
8. Перегруппировки
8.1 Перегруппировка Кляйзена
8.2 Перициклическая [3,3]-сигматропная перегруппировка
8.3 Реакция Коупа
8.4 Еноленовая перегруппировка
8.5 Перегруппировка Кляйзена аллилвиниловых эфиров
8.6 Азо-перегруппировка Коупа
8.7 [2,3] - Сигматропная перегруппировка
Список литературы
енол аллилвиниловый эфир перегруппировка
Введение
Непредельные альдегиды и кетоны, молекулы которых содержат кратные
углерод-углеродную и углерод-кислородную связи, не проявляют свойств, которые
не были бы присущи алкенам, с одной стороны, и альдегидам и кетонам - с другой.
Комбинация указанных кратных связей придает молекуле органического
соединения особые свойства в том случае, когда эти связи находятся в
сопряженном положении, т.е. когда винильный и карбонильный фрагменты
непосредственно связаны между собой.
Альдегиды и кетоны такого строения называют α,β-непредельными, поскольку кратная
связь углерод-углерод в них расположена между α- и β - углеродными атомами.
Простейшим представителем таких соединений является акролеин.
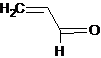
Присутствие в одной молекуле карбонильной функции и двойной связи придает
соединениям особые свойства, но только в том случае, если группы расположены
близко друг к другу. Наибольший интерес представляют кумулированные и
сопряженные системы.
Сопряжение карбонильной группы и двойной связи сильно сказывается на
спектральных свойствах, особенно на ультрафиолетовых спектрах, вследствие
стабилизации возбужденных электронных состояний, в которые при π→π* - переходах важный вклад вносят
полярные резонансные структуры.
Резонанс такого типа в основном состоянии играет значительно меньшую
роль, но выражен все же достаточно сильно для того, чтобы с его помощью можно
было объяснить сравнительно небольшое отличие дипольных моментов насыщенных и α,β-ненасыщенных альдегидов, например:
СН3СН2СНО СН2 = СН -СНО
µ = 2,7 единицы Дебая µ = 3,0 единицы Дебая
Влияние сопряжения сказывается также на частотах карбонильной группы в
ИК-спектрах и спектрах ЯМР. Что касается последних, то было найдено, что
сигналы протонов при β-углеродном атоме а,β-ненасыщенных карбонильных соединений
обычно лежат на 0,7-1,7 м. д. в более слабом поле, чем сигналы протонов обычных
олефинов. Для α-протонов этот эффект выражен слабее. [1]
α,β-Непредельные альдегиды и кетоны,
являются винилогами соответствующих альдегидов и кетонов, не содержащих
винильной группы. В них фрагмент, включающий винильную и карбонильную группы,
выступает как своеобразная единая система. Для упрощения рассмотрения
реакционной способности указанной системы все атомы в ней нумеруются:
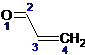
В УФ-спектрах α,β-непредельных альдегидов и кетонов,
как и в УФ-спектрах, сопряженных алкадиенов максимумы поглощения сдвинуты в
более длинноволновую область по сравнению с соответствующими насыщенными
соединениями (батохромный сдвиг), а интенсивности пиков несколько увеличены. То
же относится к производным α,β-непредельных альдегидов и кетонов. В
ряде случаев это может быть использовано в целях идентификации [2].
Химические свойства
α,β-Ненасыщенные карбонильные соединения
могут вступать в обычные реакции присоединения и конденсации по карбонильной
группе, такие, как образование циангидридов и гидразонов и присоединение
металлоорганических соединений. Эти реакции, однако, могут осложняться (если не
перекрываться полностью) 1,4 присоединением. Реактивы Гриньяра могут
взаимодействовать либо по типу 1,2-присоединения (с карбонильной), либо по типу
1,4-присоединения, в которое вовлекаются как карбонильная группа, так и двойная
связь. Условия баланса между двумя направлениями реакций настолько
чувствительны к различным воздействиям, что сравнительно небольшие изменения
характера пространственных препятствий оказываются достаточными для того, чтобы
сделать один из процессов доминирующим.
В случае цианистого водорода образование циангидрина обычно происходит
быстрее, чем 1,4-присоединение, и если положение равновесия выгодно (как и для
большинства альдегидов), то наблюдается только 1,2-присоединение.

Образование циангидринов кетонов менее выгодно, и в результате
1,4-присоединение приводит к β-цианкетону.
Присоединение галогеноводородов к α,β-ненасыщенным альдегидам и кетонам
приводит к тому, что галоген вступает в β-положение. Если винильная группа сопряжена
с карбонильной функцией, то образование продукта происходит против правила
Марковникова.
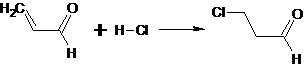
Направление присоединения к α,β-ненасыщенным кислотам имеет
аналогичный характер: продукты соответствуют реакции 1,4-типа.
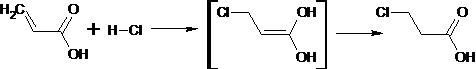
Для многих α,β-ненасыщенных альдегидов и кетонов
взаимодействие с гидразином приводит вследствие двух последовательных реакций
1,2- и 1,4-присоединения к образованию циклических продуктов, называемых
пиразолинами.
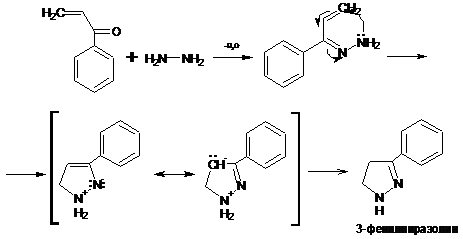
Синтез β, γ - ненасыщенных альдегидов и кетонов обычно представляет
определенные трудности; они легко перегруппировываются в α,β - непредельные изомеры, в частности,
в присутствии реагентов основного характера [24]:

Таким образом, непредельные карбонильные соединения являются важными
органическими соединениями, которые используются в синтезе ряда веществ, в том
числе и лекарственных препаратов.
1. Реакции с
участием енолов и енолят-ионов
.1
Нуклеофильное замещение с участием енолят-анионов
Енолят-анион может быть с хорошим выходом получен из кетона (если
последний содержит α-водородный атом) при действии сильных нуклеофильных
агентов, подобных амидам натрия или калия. Образовавшийся таким образом
енолят-анион теоретически может вступать в SN-реакции с алкилгалогенидами двумя
различными способами. Так, в случае трет-бутилметилкетона и йодистого метила
возможны следующие реакции, отличающиеся только положением, в которое направляется
атака:
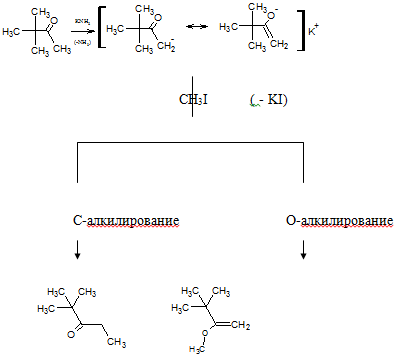
Выше в связи с альдольной конденсацией рассматривалась возможность
протекания реакций енолят-аниона таким образом, как если бы заряд был в
значительной степени сконцентрирован либо на атоме углерода, либо на атоме
кислорода. Существует, однако, глубокое различие между альдольной конденсацией
и рассматриваемыми здесь реакциями, поскольку в первом случае реакция по
кислороду, как указывалось, весьма невыгодна термодинамически по сравнению с
реакцией по углероду. В то же время и О - и С-алкилирование аниона
термодинамически выгодно. Кроме того, алкилирование в отличие от альдольного
присоединения в обычных условиях необратимо, и поэтому продукт О-алкилирования
не может перейти в продукт С-алкилирования, несмотря на то, что последний на 24
ккал более стабилен. Реакционная способность галогенида часто оказывается
определяющей для протекания алкилирования по С- и О- направлению. Решающим
фактором, по-видимому, является степень образования новой связи углеродом в
галогениде в переходном состоянии, отделяющем исходные реагенты от продуктов
реакции. Если образование новой связи произошло в значительной степени (это
обычно оказывается необходимым для того, чтобы "вытолкнуть" уходящую
группу из галогенида, реакционная способность которого в SN2-рeакциях невелика), то следует ожидать преимущественного
алкилирования по углероду. В такой ситуации переходное состояние оказывается в
большей степени сходным с продуктами реакции, и С-алкилирование соответствует
более устойчивому продукту
Если же реакционная способность галогенида велика, то образование новой
связи в переходном состоянии осуществляется в гораздо меньшей степени и
переходное состояние поэтому весьма сходно с исходными реагентами. При этом
может произойти О-алкилирование, т.к. заряд енолят-аниона в большей степени
сконцентрирован на атоме кислорода, чем на углероде [3].
.2
Конденсации енолят-анионов
.2.1
Альдольная конденсация
Енолят-анионы принимают участие во многих важных синтетических реакциях
карбонильных соединений - они либо присоединяются по двойным связям, либо
выступают в качестве атакующих агентов при нуклеофильном замещении. Если
присоединение происходит по двойной связи карбонильной группы, то эта реакция
носит название альдольной конденсации (альдольного присоединения).
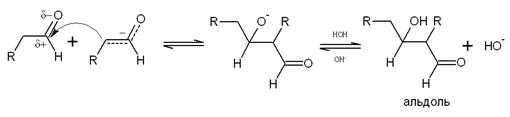
Продукт присоединения енолят-иона к альдегиду представляет собой β-гидроксиальдегид
(3-гидроксиальдегид), получивший тривиальное название альдоль (где аль -
альдегид, ол - спирт). Именно поэтому реакция получила такое название. В
классических условиях енолят-ион, принимающий участие в альдольной конденсации,
генерируется из карбонильного соединения при отщеплении протона с помощью
гидроксидиона в водной или водно-спиртовой среде при 0-5°С. Так, например,
пропаналь при обработке водным раствором гидроксида натрия при 0-5°С в течение
5 часов образует З-гидрокси-2-метилпентаналь с выходом 55-60%, а уксусный
альдегид дает альдоль с выходом 50%:
В более жестких условиях альдоли подвергаются дегидратации с образованием
α,β-ненасыщенных альдегидов, что делает
весь процесс в целом необратимым:
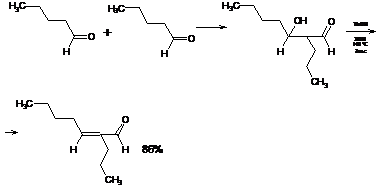
Механизм альдольной конденсации, катализируемой основанием, включает три
стадии:
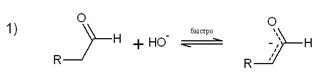
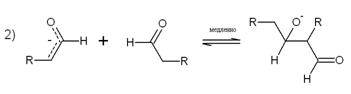
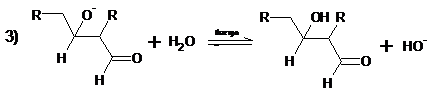
В первой стадии образуется енолят-ион. Равновесная концентрация
енолят-иона очень мала и редко превышает 1-3%, поскольку рКа
альдегида примерно равно 17-18, а рКа воды равно 15,7. Присоединение
енолят-иона к карбонильной группе неионизированной молекулы альдегида во второй
стадии определяет скорость всего процесса. Дегидратация альдоля в α,β-ненасыщенный альдегид также
катализируется основанием и протекает с промежуточным образованием енолят-иона
альдоля:
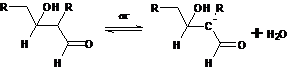
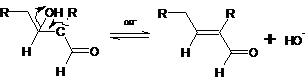
Альдольная конденсация катализируется не только основанием, но также и
кислотами. В кислой среде, как и следовало ожидать, определяющей скорость
стадией процесса является присоединение енола к карбонильной группе:
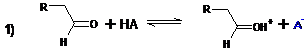

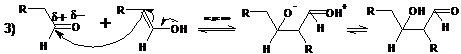
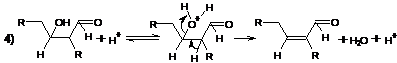
В кислой среде практически невозможно остановить реакцию на стадии
образования альдоля, и конечным продуктом оказывается α,β-ненасыщенный альдегид - продукт его
дегидратации. Для кетонов константа равновесия образования альдоля гораздо
ниже, чем для альдегидов. Для смещения равновесия в сторону альдоля разработана
специальная экспериментальная техника, которая оказалась эффективной для
простейших низкокипящих кетонов - ацетона, пентанона-3 и некоторых других
кетонов:
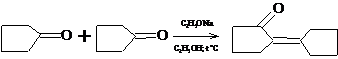
1.2.2 Внутримолекулярная циклизация дикетонов
Внутримолекулярная циклизация дикетонов представляет собой важный и
оригинальный метод синтеза циклических соединений.
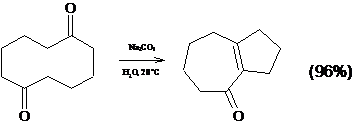
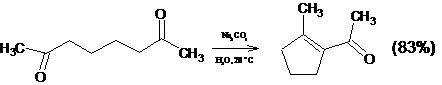
1.2.3 Перекрестная альдольная конденсация
В классических условиях альдольной конденсации в водном растворе
гидроксида натрия из смеси двух алифатических альдегидов в общем случае
получаются все четыре возможных альдоля. Чтобы перекрестная альдольная
конденсация была препаративно полезна, реакция должна быть строго селективной.
Это возможно тогда, когда один компонент выполняет роль только нуклеофильного
агента, а другой - только карбонильного акцептора. Некоторые следствия из этого
ограничения очевидны. Ароматические и гетероциклические альдегиды, так же как и
формальдегид, выполняют роль карбонильной компоненты в перекрестной альдольной
конденсации, поскольку они не содержат водорода при α-углеродном атоме:
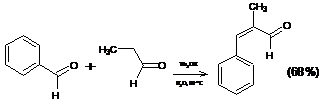
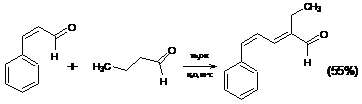
1.2.4 Реакция Кляйзена-Шмидта
Перекрестная (смешанная) альдольная конденсация ароматических альдегидов
с кетонами, приводящая к образованию α,β-ненасыщенных жирноароматических
кетонов, известна под названием реакции Кляйзена-Шмидта:
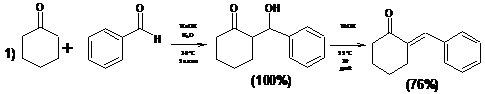
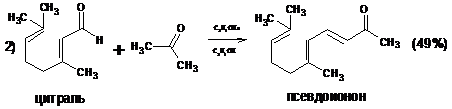
Перекрестная альдольная конденсация карбонильных соединений в протонной
среде (вода, спирт), катализируемая гидроксид- или алкоксидионом, имеет
ограниченную область применения, если конечной целью является получение самого
альдоля. Это определяется целым рядом причин. Константа равновесия для
конденсации кетонов в воде не благоприятствует образованию альдоля. Для
несимметричных кетонов в образовании альдолей принимают участие оба изомерных
енолят-иона, что приводит к образованию сложной смеси продуктов. Эта же
неопределенность сохраняется и для конденсации двух различных алифатических
альдегидов или для пары алифатический альдегид - алифатический или
алициклический кетон. Кроме того, образование альдоля в протонной среде
сопровождается дегидратацией в α,β-ненасыщенный альдегид или кетон и
скорее может рассматриваться как метод получения именно этих соединений. [4]
Характер протекания альдольной конденсации можно видеть на примере
реакции ацетальдегида с основанием, которая при проведении ее в сравнительно
мягких условиях дает (β-оксимасляный альдегид (ацетальдоль)
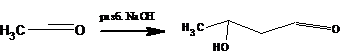
При нагревании реакционной смеси продукт дегидратируется в кротоновый
альдегид (бутен-2-аль):
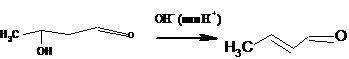
Легкость такой дегидратации (даже в щелочной среде) согласуется с
легкостью близких по характеру реакций отщепления, в которые вступают β-замещенные карбонильные соединения.
На первой стадии альдольной конденсации при отрыве α-водорода основанием образуется
енолят-анион:
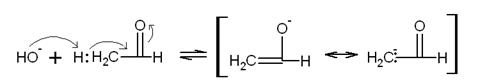
Затем анион присоединяется к карбонильной связи аналогично цианид-иону
при образовании циангидрина. Рассмотрение двух резонансных форм енолят-аниона
позволяет предположить, что присоединение может происходить по одному из двух
путей: атака аниона приводит либо к образованию углерод-углеродной связи, в
результате чего образуется альдоль II, либо к образованию связи
углерод-кислород и получению α-окси этилвинилового эфира III.
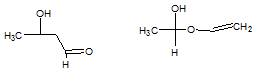
Хотя образование эфира III возможно с точки зрения механизма, оно
оказывается гораздо менее вероятным по термодинамическим соображениям.
Действительно, в то время как суммарное значение ∆Н (для газовой фазы),
вычисленное по энергиям связей, для образования альдоля равно - 4 ккал, для
образования эфира III оно составляет +20,4 ккал.
Константа равновесия при альдольной конденсации ацетальдегида имеет
благоприятное значение, так же как и для большинства других альдегидов. В
случае ацетона в равновесной смеси присутствует лишь несколько процентов
продукта присоединения - "диацетонового спирта". Это становится
понятным при учете пространственных препятствий и того факта, что карбонильная
связь в кетоне приблизительно на 3 ккал прочнее, чем в альдегиде. Несмотря на
невыгодное значение константы равновесия, диацетоновый спирт можно получить с
хорошим выходом, использовав прибор, изображенный на рис. 1.

Рис. 1. Прибор для получения диацетонового спирта
Ацетон нагревают до кипения; при этом горячий конденсат стекает из
обратного холодильника, проходит через пористую гильзу, содержащую твердую
окись бария, и устанавливается равновесие с диацетоновым спиртом. Окись бария
остается в гильзе, а жидкая фаза возвращается в колбу, где ацетон (кипящий на
110° ниже диацетонового спирта) селективно переходит в газовую фазу и
возвращается в зону реакции, чтобы вновь частично превратиться в диацетоновый
спирт.
Соединения, участвующие в ключевой стадии при альдольной конденсации,
должны быть, как правило, одно - донором пары электронов, а другое - ее
акцептором. При образовании ацетальдоля и диацетонового спирта обе роли
выполняют одинаковые молекулы, однако это вовсе не является необходимым.
Возможны смешанные реакции присоединения самых различных типов. Рассмотрим пару
формальдегид - ацетон: формальдегид не может образовать енолят-аниона, так как
у него отсутствуют α-водороды, но можно ожидать, что он окажется особенно
хорошим акцептором электронной пары. Последнее связано с тем, что он свободен
от пространственных препятствий, а также с тем, что прочность карбонильной
связи в нем необычно низка (166 ккал, ср. с 179 ккал для ацетона). Ацетон легко
образует енолят-анион, но сравнительно плохо подходит для роли акцептора.
Следовательно, присоединение ацетона к альдегиду должно происходить (и
происходит в действительности) с большой легкостью.
Особо важное значение имеет дегидратация альдолей в α,β-ненасыщенные карбонильные соединения,
протекающая наилучшим образом в условиях кислотного катализа. Другим примером
может служить дегидратация диацетонового спирта в окись мезитила [5]:
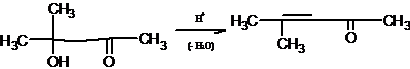
2.
Электрофильное присоединение
.1 Гидратация
Винилацетилена
В данном случае гидратация идет по тройной связи и винилацетилен
проявляет себя как алкин. Алкины, подобно алкенам, способны присоединять по
тройной связи галогены, галогеноводороды, воду. Эти реакции протекают в ряду
алкинов также по механизму АЕ. Однако алкины менее реакционноспособны
по отношению к электрофилам нежели олефины.
Это кажется на первый взгляд странным, так как алкины более ненасыщенны.
Однако все объясняется особенностями sp-гибридизованных атомов углерода: из-за их повышенной
электроотрицательности π-молекулярное облако сильнее притянуто к ядрам атомов
углерода и, как результат, труднее поляризуется. Механизм реакции АЕ у
алкинов аналогичен олефинам, однако пониженная поляризуемость затрудняет первую
стадию реакции - образование л-комплекса. Также на решающей стадии реакции
(присоединения электрофила) образуется винильный катион, менее устойчивый и
более богатый энергией в сравнении с насыщенным карбокатионом в случае алкенов.
Поэтому реакция присоединения замедляется.
Для присоединения воды к алкину недостаточно присутствия сильной кислоты
(H2SО4), как в случае алкенов. М.Г.Кучеровым было
обнаружено, что процесс облегчается в присутствии ионов двухвалентной ртути
(реакция Кучерова)[6].
Предполагают, что и в данном случае реакция начинается с протонирования
тройной связи, а образовавшийся карбокатион далее реагирует с водой как с
нуклеофилом:
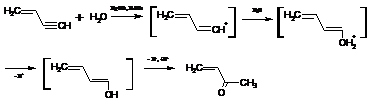
Здесь следует рассмотреть вопрос о том, почему образующийся спирт
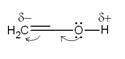
перегруппировывается в карбонильное соединение (правило Эльтекова - Эрленмейера).
Причина неустойчивости енолов обусловлена наличием - I -эффекта у винильной группы и +
М-эффекта у группы ОН, обусловленного способностью атома кислорода подавать
неподеленные пары р-электронов электроноакцепторным группам:
Следствием взаимного влияния винильной и ОН-групп является, с одной
стороны, повышение кислотности атома водорода гидроксильной группы (по аналогии
с фенолом, для которого рКа»10), а с другой - сосредоточение некоторой избыточной
электронной плотности на крайнем атоме углерода винильной группы.
Перегруппировка енола в карбонильное соединение представляется как
результат межмолекулярного взаимодействия:
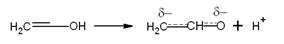
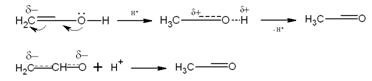
Менее вероятно, что эта перегруппировка представляет собой
внутримолекулярную миграцию протона:
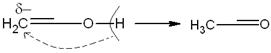
Равновесие енол ↔ карбонильное соединение у алифатических
альдегидов и кетонов практически нацело смещено вправо. Это объясняют
следующими причинами. Во-первых, такая изомеризация сопровождается выигрышем
энергии (65 кДж/моль). Следовательно, образующийся спирт, как менее устойчивый,
претерпевает перегруппировку.
Во-вторых, в результате перегруппировки образуется менее диссоциированное
соединение (рКа ацетальдегида составляет »20, енола » 10).
Возможно, каталитическое действие соли ртути заключается в том, что в
качестве интермедиата образуется ртутьорганическое соединение.
Присоединение воды к гомологам ацетилена протекает легче, присутствие ртутного
катализатора необязательно. По-видимому, смещение π-электронной плотности тройной связи
под влиянием электронодонорной алкильной группы облегчает протонирование, в
соответствии с правилом Марковникова, крайнего атома углерода на первой стадии
реакции, что подтверждает правильность предполагаемого механизма реакции
гидратации:
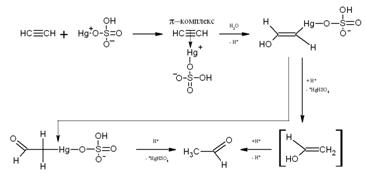
Винильная группа, имеющая помимо - I-эффекта еще и + M-эффект, делает тройную связь даже более реакционноспособной, чем
двойная. Именно поэтому при взаимодействии с водой винилацетилен образует
винилметилкетон [7]:
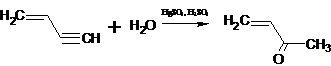
3. Нуклеофильное присоединение
Синтез α,β-непредельных карбонильных соединений
может проходить с использованием самих α,β-непредельных карбонильных
соединений. Примером таких реакций служит присоединение литийорганических
соединений.
В реакциях α,β-непредельных альдегидов и кетонов с
нуклеофильными реагентами могут затрагиваться положения 2 и 4 сопряженной
системы, что объясняется эффективностью передачи полярного влияния карбонильной
группы через винильную. Таким образом, нуклеофил в обоих случаях атакует
электронодефицитные атомы углерода. Стабилизация образовавшегося аниона
осуществляется присоединением противоиона к атому кислорода, т.е. по положению
1. В общем виде схема превращений обоих типов выглядит следующим образом (Nu-нуклеофил, А-противоион):
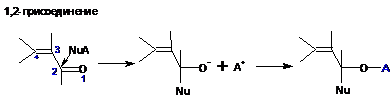
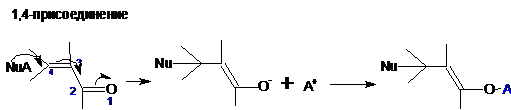
Продукты 1,4-присоединения при А=Н неустойчивы и как другие виниловые спирты
переходят в соответствующие альдегиды или кетоны:
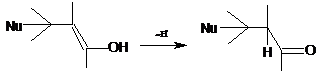
Пока еще не найдены общие закономерности, которые бы ни позволили
предсказать, по какому пути в том или ином конкретном случае пойдет
присоединение. Считается, что α,β-непредельные альдегиды более склонны
к 1,2-присоединению, а α,β-непредельные кетоны к
1,4-присоединению. Эти тенденции наблюдаются в реакциях с самыми различными
нуклеофилами.
Из реакций α,β-непредельных альдегидов и кетонов с
сильными нуклеофилами важное значение имеет взаимодействие с
металлоорганическими соединениями.
Показано, что магнийорганические соединения присоединяются α,β-непредельным альдегидам почти
исключительно по 1,2-положениям. а к кетонам - преимущественно по
1,4-положениям.
Применение же литийорганических соединений дает возможность получать
продукты 1,2-присоединения (т.е. соответствующие спирты) в обоих случаях.
В то же время α,β-непредельные карбонильные соединения
независимо от строения взаимодействуют с литийалкилкупратами только с
образованием продуктов 1,4-присоединения, что используется для получения
альдегидов и кетонов с усложненным углеродным скелетом из продуктов кротоновой
конденсации [8]:
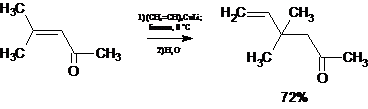
4. Реакции
отщепления
Реакциями отщепления или элиминации называются реакции, при которых из
молекулы удаляются два атома или две группы без замещения другими атомами или
группами. В большинстве таких реакций атомы или группы уходят от
двух соседних атомов углерода (причем очень часто от одного из них отщепляется
протон, а от другого нуклеофил А: или А-), и между этими атомами
углерода образуется кратная связь:

Хорошо известными примерами реакций отщепления могут служить:
катализируемое основаниями отщепление галогеноводорода от алкилгалогенидов,
катализируемая кислотами дегидратация спиртов, а также разложение гидроокисей
четвертичных алкиламмоний-производных по Гофману.
Механизм Е1.
При механизме Е1, так же как и в случае механизма SN1, скорость реакции зависит только от
концентрации субстрата, т. е. в стадии, лимитирующей скорость такой реакции,
участвуют только молекулы субстрата. Так, в случае реакций с участием
галогенида Ме3СВг
Скорость ~ [Ме3СВr]
измеряемая скорость есть по существу скорость образования переходного
состояния, моделью которого может служить карбониевый ион, входящий в состав
ионной пары:
Me3CBr → Ме3С+ Вr-
За этой стадией может следовать быстро протекающая и поэтому не
лимитирующая скорость реакции атака другими компонентами системы, например
ионами _ОН или молекулами Н2O. Если эти атакующие агенты действуют как нуклеофилы (т. е.
как доноры электронной пары по отношению к углеродному атому), то суммарным результатом
реакции будет замещение:
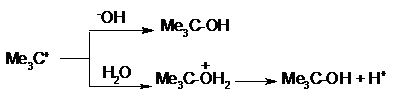
Если же указанные агенты действуют как основания (т. е. как доноры
электронной пары по отношению к водороду), то в этом случае результатом реакции
будет отщепление протона от β-углеродного атома; с образованием
алкена:
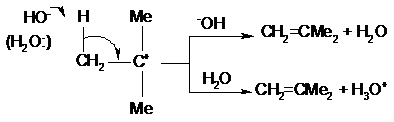
Очевидно, что условия, способствующие реакциям SNl, должны ускорять также и реакции E1, поскольку в обоих случаях
существенной стадией является образование карбониевого иона. Действительно,
было показано, что отношение мономолекулярного отщепления к замещению
сохраняется обычно почти постоянным для данной алкильной группы независимо от
того, атом какого галогена (или какая другая группа) отщепляется в виде аниона.
Отсюда следует: 1) что реакции E1 и SN1 не являются совершенно
независимыми; 2) что эти реакции имеют конкурирующие механизмы и 3) что общим
промежуточным соединением для них является карбониевый ион (в противном случае
соотношение продуктов замещения и отщепления существенно менялось бы в
зависимости от природы уходящей группы). Изменение структуры алкильной группы
оказывает, однако, значительное влияние на соотношение замещения и отщепления.
Показано, что разветвление при β-углеродном атоме способствует
отщеплению по механизму E1.
Так, в случае МеCН2СМе2Сl образуется только 34% алкена, тогда
как в случае Ме2СНСМе2Сl - уже 62%. Такой эффект, по крайней мере частично, можно
объяснить стерическими причинами: чем более разветвленными являются молекулы
галогенида, тем более "сжатыми" они оказываются при превращении в
промежуточную структуру - карбониевый ион. "Сжатие" возрастает, когда
карбониевый ион взаимодействует со вступающей группой (это приводит к
замещению), и, наоборот, уменьшается, если происходит потеря протона, что делает
отщепление более предпочтительным. Изучение реакций ряда галогенидов
показывает, однако, что подобное объяснение не является исчерпывающим. При
предпочтительном образовании одного из двух изомерных алкенов из карбониевого
иона, в котором имеется более чем один β-атом углерода, способный терять
протон, существенное значение могут иметь и такие эффекты как сверхсопряжение.
По механизму E1 протекает также
дегидратация спиртов, катализируемая кислотами [9]:
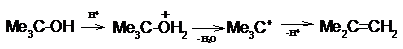
4.1 Дегидратация
спиртов
.1.1
Механизмы дегидратации спиртов
Протонирование спиртов в ненуклеофильной среде приводит к дегидратации,
которая происходит при нагревании спирта в концентрированной серной, фосфорной
кислотах или в суперкислой среде - смеси пятифтористой сурьмы и фторсульфоновой
кислоты. Катион алкоксония, отщепляя воду, образует нестабильный интермедиат-
карбокатион, который теряет протон с образованием алкена. Наиболее медленная
стадия всего процесса - превращение катиона алкоксония в карбокатион. Концентрированная
H2SO4 или H3PO4 связывают
выделяющуюся воду, что делает весь процесс необратимым.
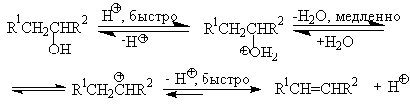
Приведенная последовательность превращений типична для реакций
мономолекулярного элиминирования Е1, продукты которого определяются правилом
Зайцева, т.е. преобладает наиболее разветвленный при двойной связи алкен.
Вторичные спирты подвергаются дегидратации при нагревании с 85%-ной
Фосфорной кислотой при 160-170оС или с 60-70%-ной серной
кислотой при 90-100оС и направление дегидратации соответствует
правилу Зайцева.
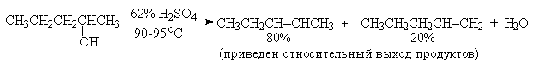
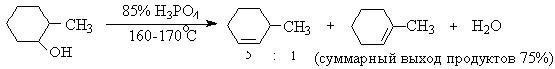
Дегидратацию третичных спиртов можно проводить уже в 20-50%-ной серной
кислоте при 85-100 oС.
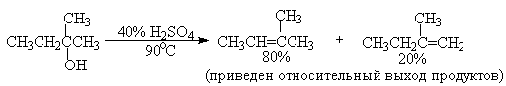
Третичные спирты дегидратируются так легко, что возможна избирательная
дегидратация диола, содержащего третичную и первичную гидроксильные группы.
Для E1-элиминирования, также как и для других процессов с образованием
карбокатиона в качестве интермедиата, характерны перегруппировки, включающие
анионотропную 1,2-миграцию гидрид-иона или алкильной группы. В качестве примера
можно привести кислотно-катализируемую дегидратацию 3-метилбутанола-2 в 80%-ной
серной кислоте.
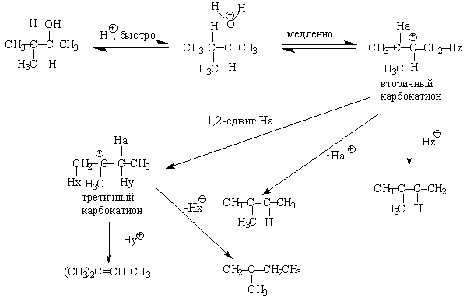
Примером скелетной изомеризации карбокатиона в Е1-элиминировании может
служить дегидратация 3,3-диметилбутанола-2.

Для первичных спиртов, вероятно, реализуется иной, Е2 механизм
дегидратации в концентрированной серной кислоте, они дегидратируются в гораздо
более жестких условиях. Так, пропанол-1 дает пропилен при нагревании с 96%-ной
серной кислотой при 170-190oС, этанол в этих же условиях дает
этилен.
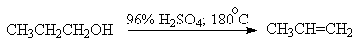
Первичные спирты при взаимодействии с серной кислотой легко образуют
полуэфиры серной кислоты. Е2-Элиминированию в этом случае, по-видимому, подвергается
полуэфир, а роль основания выполняет гидросульфат-ион или вода.
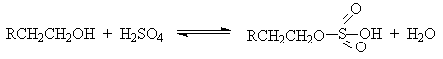
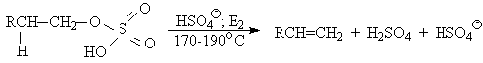
Возможен и другой механизм дегидратации первичных спиртов, в котором
субстратом является катион алкоксония, а основанием гидросульфат-ион.
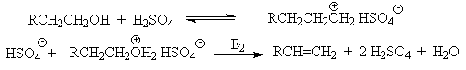
В более мягких условиях при нагревании простейших первичных спиртов с
96%-ной серной кислотой при 130-140oС преимущественно получаются
простые эфиры. При этом первичный спирт алкилируется либо под действием
полуэфира серной кислоты, либо при взаимодействии с катионом алкоксония.
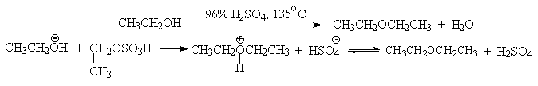
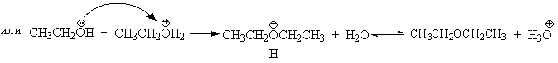
Этим способом получают простейшие простые эфиры - диэтиловый,
дипропиловый и дибутиловый эфиры и циклические простые эфиры, например,
тетрагидрофуран и диоксан. Вторичные и третичные спирты в этих условиях
дегидратируются с образованием алкенов.
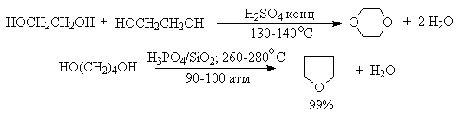
Этот способ неприемлем для получения несимметричных эфиров из двух
спиртов, так как при этом образуется смесь трех возможных продуктов ROR, R'OR,
R'OR'.
В промышленности для внутри- или межмолекулярной дегидратации вместо
серной кислоты в качестве дегидратирующего средства используют безводную окись
алюминия. Гетерогенная каталитическая дегидратация первичных, вторичных и
третичных спиртов над окисью алюминия при 350-450oС приводит к
алкенам [21].
4.1.2
Дегидратация глицерина в присутствии гидросульфата калия
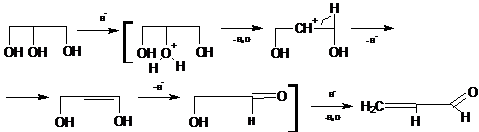
В лаборатории акролеин получается дегидратацией глицерина в присутствии
гидросульфата калия. При нагревании глицерина в присутствии водоотнимающих
средств образуется акролеин - непредельный альдегид, имеющий запах кухонного
газа. Это одна из качественных реакций на глицерин.
Глицерин при перегонке над бисульфатом калия отщепляет две молекулы воды,
в результате чего образуется непредельный альдегид - акролеин[10]:
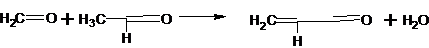
Ранее в промышленности был распространён процесс парофазной кротоновой
конденсации ацетальдегида с формальдегидом (устаревший метод):
В настоящее время акролеин получают каталитическим
окислением пропилена над оксидными висмут-молибденовыми катализаторами или
оксидом меди. Этот метод и его механизм рассмотрены в следующем разделе (см.
каталитическое окисление алкенов).
5. Реакции
окисления
.1 Окисление
первичных и вторичных спиртов
Для окисления первичных и вторичных спиртов соответственно до альдегидов
и кетонов применяют многие из обычных окислителей - смесь дихроматов натрия
(смесь Килиани) или калия (смесь Бекмана) с серной кислотой, триоксид хрома,
перманганат калия, диоксид марганца.
Для окисления первичных спиртов до альдегидов применяют смесь
концентрированной серной кислоты и дихромата натрия; образующийся альдегид во
избежание дальнейшего окисления отгоняют из реакционной смеси. Это возможно,
так как получающиеся альдегиды как неспособные к ассоциации кипят значительно
ниже соответствующих спиртов:
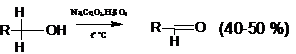
Хорошие результаты дает также окисление первичных спиртов оксидом хрома(VI) в сухом пиридине. Также в последнее
время в промышленности первичные спирты стали окислять до альдегидов кислородом
воздуха в присутствии катализаторов [11].
.1.1 Окисление
первичных и вторичных спиртов с помощью ДМСО
В последние тридцать лет разработано несколько эффективных способов
окисления первичных и вторичных спиртов с помощью ДМСО или комплексов ДМСО с
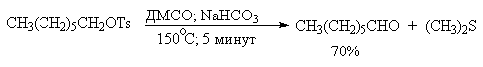
электрофильными агентами. Тозилаты первичных спиртов, также как и
бензилтозилаты, окисляются в альдегиды при нагревании в ДМСО в течение 10-30
минут при 120-150оС в присутствии гидрокарбоната натрия как слабого
основания.
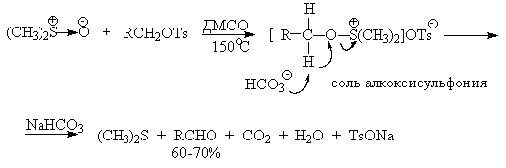
ДМСО в этой реакции выполняет роль нуклеофильного агента, который
замещает тозилоксигруппу по обычному SN2 механизму с образованием
алкоксисульфониевой соли. Катион алкоксисульфониевой соли далее подвергается
окислительно-восстановительному элиминированию по механизму, аналогичному для
окислительно-восстановительного элиминирования из сложных эфиров хромовой
кислоты. Гидрокарбонат-ион является основанием в этой Е2 реакции
элиминирования, приводящей к диметилсульфиду и альдегиду. В качестве примера
приведем получение гептаналя и п-бромбензальдегида.
Слабый нуклеофильный агент ДМСО легко превращается в сильный
электрофильный агент, который реагирует со спиртами уже ниже 0oС в
мягких условиях. Необходимую активацию ДМСО проводят с помощью серного
ангидрида, трифторуксусного ангидрида, N-хлорсукцинимида или
N,N-дициклогексилкарбодиимида C6H11N=C=NC6H11
(ДЦГК). Во всех случаях в качестве реакционноспособного интермедиата
образуется активированная алкоксисульфониевая соль, которая далее подвергается
внутримолекулярной окислительно-восстановительной фрагментации.
Для окисления первичных и вторичных спиртов до альдегидов и кетонов в
мягких условиях эффективен комплекс ДМСО с SO3, образующийся при
взаимодействии пиридинсульфотриоксида с ДМСО.
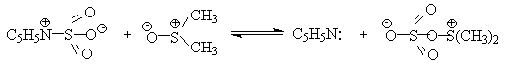
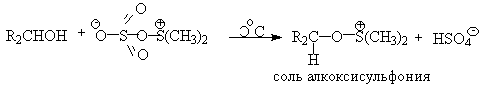
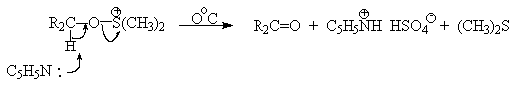
SO3 в качестве электрофильной частицы может быть заменен
трифторуксусным ангидридом или ДЦГК (реактив Пфитцера-Моффата). Этот реагент в
настоящее время употребляется наиболее часто.
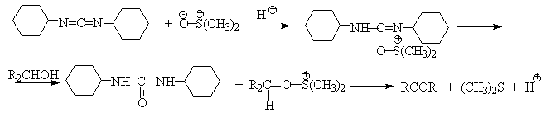
Пример окисления спиртов комплексами ДМСО:
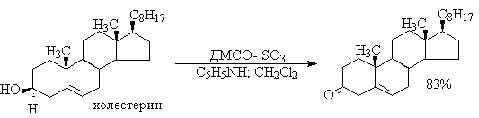
5.1.2 Окисление вторичных спиртов по Оппенауэру
Эти методы окисления вытеснили старый громоздкий способ окисления
вторичных спиртов по Оппенауэру, который заключается в нагревании спирта с
алкоголятом алюминия в присутствии карбонильного соединения в качестве
акцептора гидрид-ионов. Этот процесс обратим (обратная реакция называется
восстановлением по Меервейну-Понндорфу-Верлею). Равновесие можно сместить
вправо, если выбрать сильный акцептор гидрид-иона - п-хинон, бензофенон,
хлоранил (2,3,5,6-тетрахлор-1,4-бензохинон).
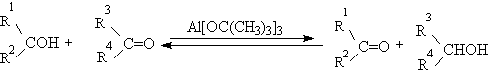
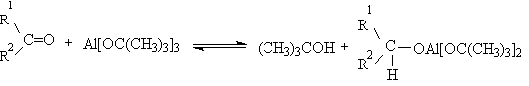
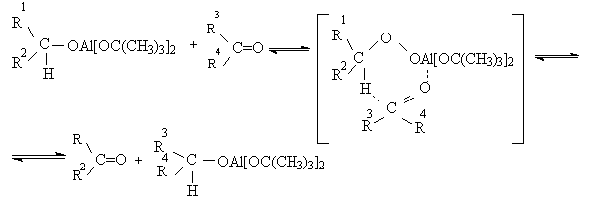
Окисление спиртов по Оппенауэру в теоретическом отношении представляет
собой пример окислительного процесса с переносом гидрид-иона от восстановителя
к окислителю в одну стадию, в то время, как в выше описанных процессах
окисление спиртов осуществляется в несколько стадий с последовательным
переносом одного или нескольких электронов [12], [25].
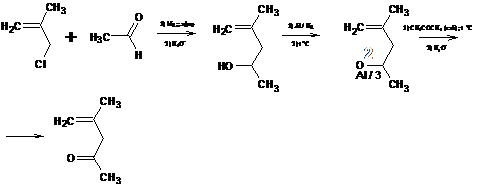
5.2 Окисление одноатомных непредельных спиртов (алкенолы и алкинолы)
Непредельные спирты (алкенолы и алкинолы) - производные непредельных
углеводородов, в молекулах которых водородный атом замещен на гидроксильную
группу. Такие спирты имеют различное строение. В одном случае гидроксильная
группа находится непосредственно при углеродном атоме с двойной связью (I), в
другом - при углероде, не связанном двойной связью (II):

Спирты типа (I) - этиленовые спирты - неустойчивы и в свободном состоянии
не встречаются. В растворах они могут существовать непродолжительное время,
изомеризуясь затем в более стабильную форму - альдегиды или кетоны (правило А.
Л. Элътекова).
Окисление первичных спиртов в альдегиды и вторичных спиртов в кетоны
является одним из важнейших превращений функциональных групп и оценкой
избирательного действия реагента, используемого в качестве окислителя.
Третичные спирты не окисляются, а в жестких условиях окисление
сопровождается деструкцией углеродного скелета. Наиболее широкое применение для
окисления спиртов нашли реагенты на основе переходных металлов - хрома (VI),
марганца (VII), марганца (IV).
Окисление алкенола:
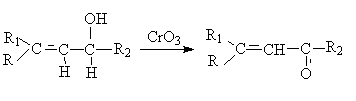
Окисление алкинола:
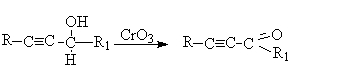
Лучшими реагентами для окисления первичных спиртов в альдегиды являются
комплекс CrO3 с двумя молями пиридина (реагент Саррета-Коллинза) и
хлорхромат пиридиния C5H5N+H.CrO3.Cl-
(реагент Кори) в хлористом метилене. Реагент Саррета-Коллинза получается при
медленном введении оксида хрома (VI) к пиридину при 10-15оС.
Оранжевый комплекс CrO3c пиридином и HСl получается при добавлении
пиридина к раствору CrO3 в 20%-ной соляной кислоте. Оба эти реагента
растворимы в CH2Cl2 или CHCl3.
Ниже приведены некоторые наиболее типичные примеры окисления первичных
спиртов до альдегидов комплексами оксида хрома VI.
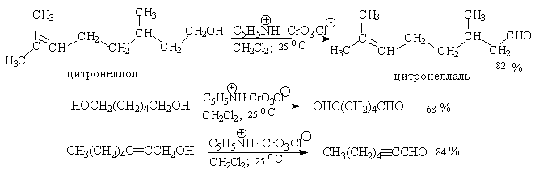
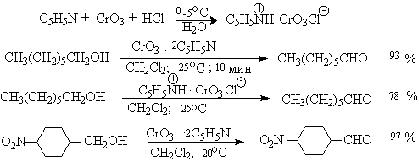
Оба окислителя обеспечивают очень высокие выходы альдегидов, однако
хлорхромат пиридиния имеет важное преимущество, так как он не затрагивает
двойную и тройную связи и может быть использован для получения ненасыщенных альдегидов.
Для получения α,β-ненасыщенных альдегидов окислением
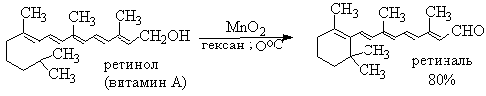
замещенных аллиловых спиртов универсальным окислителем является оксид марганца
(IV) MnO2. Этот реагент окисляет в петролейном эфире или хлористом
метилене ненасыщенные спирты с одной или несколькими двойными или тройными
связями без изомеризации и перегруппировки, что с успехом используется в
синтезе природных соединений.
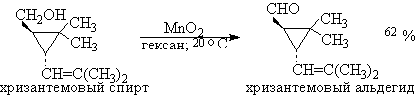
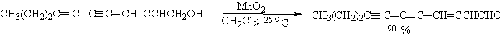
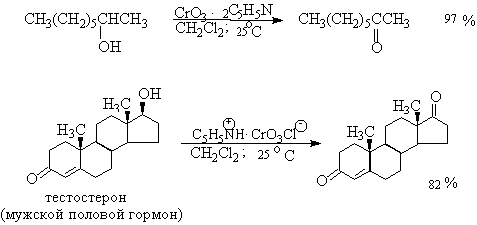
Комплексы хромового ангидрида с пиридином окисляют и вторичные спирты до
кетонов с почти количественными выходами.
Однако чаще всего для окисления вторичных спиртов используют реактив
Джонса - раствор строго рассчитанного количества CrO3 в водной
серной кислоте. Важное достоинство реагента Джонса состоит в том, что вторичные
спирты, содержащие двойную или тройную связь, быстро окисляются до кетонов без
затрагивания кратных связей.
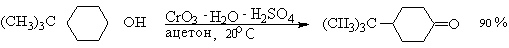
Первичные спирты окисляются реактивом Джонса до карбоновых кислот.
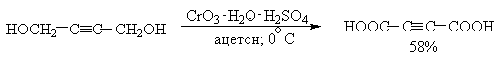
Механизм окисления спиртов под действием хромового ангидрида подробно
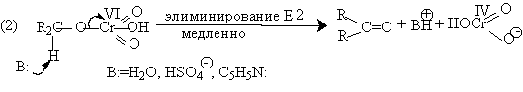
изучен. Эта реакция включает несколько стадий. Сначала из спирта и CrO3
образуется сложный эфир хромовой кислоты. Во второй, ключевой, стадии имеет
место окислительно-восстановительное элиминирование, приводящее к образованию
альдегида или кетона и частицы, содержащей Cr(IV). При окислении
дейтерированного CH3CD(OH)CH3 и недейтерированного пропанола-2
наблюдается кинетический изотопный эффект КН/KD=7. Столь
значительный первичный кинетический изотопный эффект показывает, что
элиминирование является наиболее медленной стадией, определяющей скорость всего
процесса [22]:
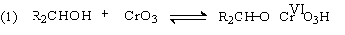
Установлено, что частицы, содержащие хром (IV), также принимают участие в
окислении спирта [13]:
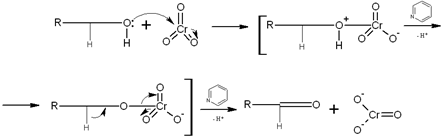
Окисление под действием соединений Cr (IV) можно полностью подавить с
помощью солей, содержащих ионы Mn (II) или Ce (III), которые окисляются Cr
(IV). Каталитические количества Ce (IV) также подавляют эту побочную реакцию,
поскольку Ce (IV) катализируют диспропорционирование Cr(IV) на Cr(III) и Cr
(VI) [23].
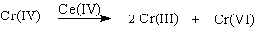
6.
Свободнорадикальное замещение
.1
Каталитическое окисление алкенов
Реакции окисления алкенов целесообразно подразделить на две большие
группы: реакции, в которых сохраняется углеродный скелет и реакции
окислительной деструкции углеродного скелета молекулы по двойной связи. К
первой группе реакций относятся эпоксидирование, а также гидроксилирование,
приводящее к образованию вицинальных диолов (гликолей). В случае циклических
алкенов при гидроксилировании образуются вицинальные транс- или цис-диолы.
Другая группа включает озонолиз и реакции исчерпывающего окисления алкенов,
приводящие к образованию различного рода карбонильных соединений и карбоновых
кислот [12].
Реакции алкенов, не сопровождающиеся разрывом двойной углерод-углеродной
связи, менее распространены, чем реакции присоединения. Чаще всего они
протекают таким образом, что гомолитически разрывается С-Н-связь, образованная
атомом углерода, соседним с винильной группой. В простейшем случае, когда
алкеном является пропен, в результате такого разрыва, который, как оказалось,
требует существенно меньшей затраты энергии (323 кДж/моль), чем разрыв
С-Н-связи при третичном атоме углерода (384 кДж/моль) в алканах, образуется
аллильный радикал:
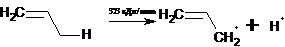
Этот радикал, взаимодействуя с реагентом, дает продукт так называемого
"аллильного" замещения общей формулы СН2=СН-СН2Х.
Легкость образования аллильного радикала, а следовательно, "легкость
свободнорадикального замещения атома водорода в аллильном положении объясняются
тем, что винильная группа способна эффективно участвовать в делокализации
неспаренного электрона на связанном с ней атоме углерода (часто говорят:
"в делокализации спиновой плотности") за счет смещения подвижных
электронов π-связи.
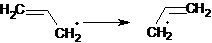
Формулы, отображающие случаи предельно возможных смешений электронов,
будут выглядеть для аллильного радикала следующим образом [14]:
Как отмечалось ранее, в промышленности акролеин получают каталитическим
окислением пропилена над оксидными висмут-молибденовыми катализаторами или
оксидом меди. Данная реакция служит примером каталитического окисления алкенов.
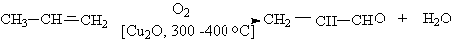
Насколько можно судить из патентных данных, в присутствии катализаторов,
приготовленных на основе молибдата висмута или оксида сурьмы, сначала из
пропилена генерируется аллильный радикал, который в результате окисления и
дальнейших превращений дает акролеин:
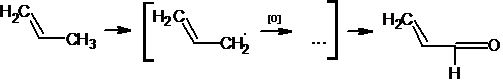
Возможность окисления "аллильного" атома углерода в алкенах с
сохранением двойной углерод-углеродной связи и получения таким путем
непредельных кетонов подтверждена экспериментально. Соответствующее превращение
(выход кетона составляет от 20 до 80%) осуществляют, обрабатывая алкен
пероксидом трет-бутила в ацетонитриле в присутствии гексакарбонила хрома в
качестве катализатора[15]:
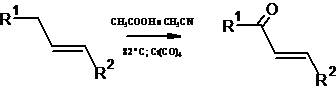
7. Перициклические
реакции
7.1
Диеновый синтез
Большое значение имеет способность α,β-непредельных альдегидов и кетонов
вступать в реакции диенового синтеза. Суть превращения, которое, относится к
классу перициклических реакций, состоит в том, что винильный фрагмент α,β-непредельного карбонильного
соединения, выступая в качестве диенофила, присоединяется по 1,4-положениям
диеновой системы с образованием соединения ряда циклогексена:

В этом случае диенофил является акцептором электронов и введение
карбонильной группы в его винильный фрагмент облегчает реакцию. Увеличение же
степени замещенности винильного фрагмента ингибирует ее. Например, оксид
мезитила в диеновый синтез обычно не вступает.
Конфигурация диенофила сохраняется в полученном аддукте. Это
свидетельствует о том, что присоединение происходит одновременно по обоим
концам реагентов. В образующемся реакционном комплексе диен и диенофил
располагаются таким образом, чтобы происходило максимальное перекрывание
орбиталей кратных связей.
Из α,β-непредельных альдегидов и кетонов в реакциях диенового
синтеза чаще других используют акролеин и его гомологи, а также метилвинилкетон
[16]:
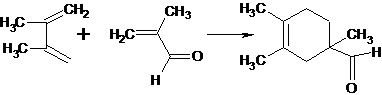
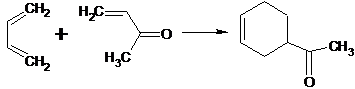
α,β-Ненасыщенные альдегиды, например
акролеин, способны вступать в реакцию Дильса-Альдера, причем они могут
реагировать не только как диенофилы, но и как диены:
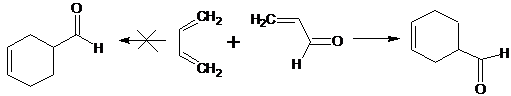
На основании этого экспериментального факта было сформулировано правило
"наибольшей кислородной плотности", согласно которому участники
реакции в начальный момент ориентируются друг относительно друга так, чтобы
атомы кислорода находились возможно ближе друг к другу. Аналогично реагируют
кротоновый альдегид и винилметилкетон. [17]
Как установил С.В.Лебедев, дивинил димеризуется при нагревании до 150°С.
Это превращение, как и димеризация алленов, является примером реакции
циклоприсоединения. При этом одна молекула дивинила реагирует, будучи в
"цисоидной" s-цис-конформации
по положениям 1 и 4, а вторая - в "трансоидной" s-транс-конформации по положениям 1 и
2. Первая выступает в качестве диена, а вторая - в качестве диенофила; продукт
реакции называют аддуктом.
Подобные реакции были подробно изучены Дильсом и Альдером и получили
название диенового синтеза. Диеновый синтез стал одним из важнейших методов
получения циклических соединений. В качестве диенофилов используют широкий
ассортимент этиленовых соединений, обычно содержащих электроноакцепторные
заместители в винильном фрагменте. Оказалось, что эта реакция не сопровождается
образованием каких-либо промежуточных частиц и что конфигурация диенофила
сохраняется в аддукте:
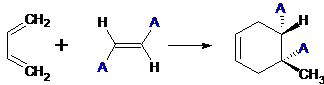
Поскольку в рассматриваемой реакции система из четырех π-электронов взаимодействует с системой
из двух π-электронов, ее называют "4 +
2-циклоприсоединением". Механизм таких реакций долгое время был не ясен и
их называли реакциями "без механизма": имевшиеся экспериментальные
данные свидетельствовали лишь о том, что в процессе превращения образуются
комплексы (иногда их называли "ионными"), в которых перемещение
кратных связей происходит синхронно или, как говорят, согласованно.
В 1965 г. Вудвард и Гофман опубликовали серию работ, в которых изложили
теорию, позволяющую предсказать для каждого конкретного случая, низкую или
высокую энергию активации будет иметь реакция, если ее инициировать нагреванием
или УФ-облучением. Низкая энергия активации, как показали авторы, присуща лишь
разрешенным процессам, т.е. тем, которые протекают через стадию образования реакционных
комплексов, в которых достигается максимальное связывание.
Последнее, как уже отмечалось, осуществляется в результате взаимодействия
π-орбиталей, которые обычно изображают
в виде двух долей, каждой из которых придают знак "+" или
"-", обозначая тем самым, что находящиеся на них электроны могут быть
в фазе или в противофазе. При сближении долей орбиталей с одинаковыми знаками
происходит связывание, а с противоположными - разрыхляющее взаимодействие.

Следовательно, низкую энергию активации должны иметь процессы, при
которых сближаются доли орбиталей с одинаковыми знаками.
Было установлено, что энергетика рассматриваемых реакций определяется
взаимодействием, так называемых граничных молекулярных орбиталей - верхней занятой
(ВЗМО) и нижней свободной (НСМО) (Следует иметь в виду, что помимо диенового
синтеза и других реакций циклоприсоединения сказанное относится также к
превращениям, которые протекают синхронно через стадию образования циклических
переходных состояний и которые объединяют под общим названием
"перициклические реакции").
Принимая во внимание все изложенное выше, построим ВЗМО и НСМО для
молекул этилена и бутадиена. В этилене взаимодействуют две атомные орбитали
углерода и в соответствии с этим возникают две молекулярные орбитали -
связывающая (доли обеих орбиталей взаимодействуют в фазе) и разрыхляющая
(взаимодействие осуществляется в противофазе). Связывающая орбиталь ψ1 несет два электрона, а разрыхляющая орбиталь ψ2 свободна:
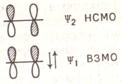

Следовательно, для этилена ВЗМО будет орбиталь у, а НСМО - орбиталь ψ2. Орбиталь ψ1 симметрична относительно плоскости, проходящей через
середину межъядерной лини перпендикулярно ей. Орбиталь ψ2 антисимметрична относительно той же плоскости и
содержит один узел (место изменения знака соседней орбитали).
Аналогичный подход применяют к построению молекулярных орбиталей
бутадиена: их должно быть четыре (взаимодействуют четыре атомные орбитали) -
две связывающие и две разрыхляющие:

Орбитали ψ1 и ψ2 - связывающие: ψ1 - симметрична и не имеет узлов, ψ2, являющаяся ВЗМО, - антисимметрична и имеет один
узел. Разрыхляющая орбиталь ψ3, являющаяся НСМО,- симметрична и имеет два узла, а
разрыхляющая орбиталь ψ4 антисимметрична и имеет три узла.
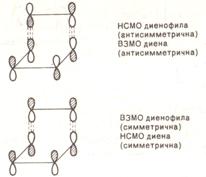
При диеновом синтезе могут реализоваться два варианта, когда во
взаимодействии участвуют: НСМО этиленового соединения (диенофила) и ВЗМО диена,
в этом случае диенофил является акцептором электронов, а диен - донором; ВЗМО
диенофила и НСМО диена, в этом случае диенофил является донором электронов:
Таким образом, в процессе диенового синтеза взаимодействуют орбитали
одинаковой симметрии.
Поскольку при формировании перициклического переходного состояния
одновременно фиксируются оба конца диенофила, конфигурация его сохраняется в
продукте реакции [18].
8.
Перегруппировки
8.1
Перегруппировка Кляйзена
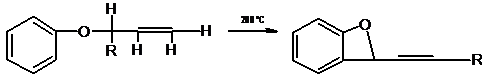
Аллилариловые эфиры при нагревании перегруппировываются в о-аллилфенолы;
эта реакция носит название перегруппировки Кляйзена. Если в исходном соединении
оба орто-положения заняты, аллильная группа мигрирует в пара-положение (в этом
случае процесс часто называют пара-перегруппировкой Кляйзена). Иногда
пара-замещенные продукты получаются и из тех эфиров, в которых одно или даже
оба орто-положения свободны, однако в целом можно сказать, что когда одно или
оба орто-положения не заняты, продуктом будет о-аллилфенол, а когда заняты оба
орто-положения. продуктом будет пара-соединение. Если замещены пара- и оба
орто-положения, реакция не идет. Миграция в мета-положения не наблюдалась. При
миграции в орто-положенне аллильная группа всегда претерпевает аллильный сдвиг,
т. е. заместитель в α-положении к кислороду оказывается в γ-положении к кольцу, и наоборот (как
показано в приведенной выше схеме). При миграции в пара-положение аллильного
сдвига не происходит, и аллильная группа сохраняет ту же структуру, что и в исходном
эфире. Пропаргильные группы (т. е. группы, содержащие тронную связь в
соответствующем положении), как правило, в эту реакцию не вступают.
8.2 Перициклическая
[3,3]-сигматропная перегруппировка
Все эти данные соответствуют механизму согласованной перициклической
[3,3]-сигматропной перегруппировки. Для орто-перегруппировки механизм имеет
вид:
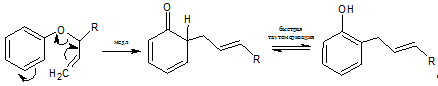
В пользу этого механизма свидетельствуют следующие данные: отсутствие
необходимости в катализаторе, кинетика первого порядка по исходному эфиру,
отсутствие продуктов перекрестной миграции при нагревании смесей, наличие
аллильного сдвига. Аллильный сдвиг при орто-перегруппировке (и отсутствие его в
пара-перегруппировке) подтверждают данные эксперимента с соединениями, меченными
изотопом 14С; эта закономерность сохраняется даже в отсутствие
заместителей в бензольном кольце исходного эфира.
.3
Реакция Коупа
Показано, что предпочтительной геометрией переходного состояния
перегруппировки Кляйзена является, как и в перегруппировке Коупа, форма кресла.
Если в орто-положениях нет атомов водорода, за перегруппировкой Кляйзена
следует вторая [3,3]-сигматропная миграция (реакция Коупа):
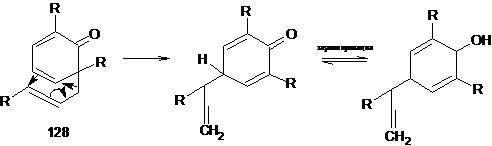
и мигрирующая группа восстанавливает свою первоначальную структуру.
Интермедиат 128 улавливают с помощью реакции Дильса - Альдера.
Эфиры с алкильной группой в γ-положепни (системы ArO-С-С=С-R) иногда дают аномальные продукты, в которых атом углерода из
β-положения присоединяется к
бензольному кольцу:

8.4
Еноленовая
перегруппировка
Установлено, что аномальные продукты не возникают непосредственно из
исходных эфиров, а образуются в результате дальнейшей перегруппировки обычного
продукта:
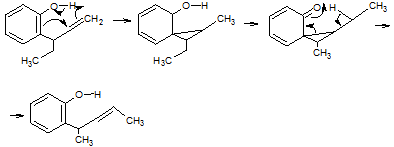
Эту перегруппировку называют еноленовой перегруппировкой, гомодиенильным
[1,5]-сигматропным сдвигом водорода и [1,5]-гомосигматропной перегруппировкой.
Она заключается в сдвиге трех электронных пар через семь атомов. Найдено, что
эта "аномальная" перегруппировка Кляйзена носит общий характер; в
результате этой перегруппировки происходит взаимопревращение енольных форм в
системах типа 129 и 131, при этом в качестве интермедиата образуется замещенный
циклопропан 130.
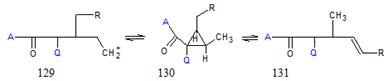
А = Н, R, Ar, OR и т.д. Q = H, R, Ar, COR, COAr, COOR и т.д.
Поскольку механизм перегруппировки Кляйзена не включает ионы, наличие или
отсутствие заместителей в бензольном кольце не должно оказывать большого
влияния на реакцию. Действительно, электронодонорные группы повышают, а
электроноакцепторные понижают скорость реакции, но эффект этого невелик; так,
п-аминосоединеиня реагируют лишь в 10-20 раз быстрее, чем п-питропроизводные.
Однако эффекты растворителя выражены значительно сильнее: при проведении
реакции в 17 различных растворителях скорость может меняться более чем в 300
раз. Наиболее подходящим растворителем является трифтороуксусная кислота, в
этом растворителе реакция идет при комнатной температуре. Перегруппировки
Кляйзена чаше всего проводят без катализатора, но иногда используют A1Cl3 или ВF3. В этом случае может происходить
реакция Фриделя - Крафтса, механизм которой не включает образование цикла и
могут получаться орто-, мета- и пара-продукты.
8.5
Перегруппировка Кляйзена
аллилвиниловых эфиров
Аллиловые эфиры енолов (аллилвиниловые эфиры) также подвергаются
перегруппировке Кляйзена; фактически перегруппировка была открыта сначала на
этих соединениях:
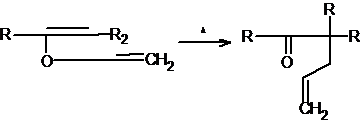
В данном случае заключительная таутомеризация. конечно, не наблюдается
даже при R'= H, поскольку здесь отсутствует необходимость восстановления
ароматичности, а кетоны более устойчивы, чем енолы. Механизм аналогичен
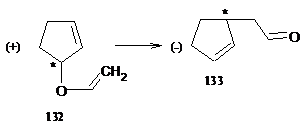
механизму перегруппировки аллилариловых эфиров. Это подтверждено, в частности,
превращением оптически активного эфира 132 в соединение 133. сохраняющее
оптическую активность. Одновременно это превращение служит еще одним примером
асимметрической индукции:
При обработке кетонов аллиловым спиртом в присутствии кислотного
катализатора можно получить γ,δ-ненасыщенные кетоны; по-видимому,
процесс идет через начальное образование виниловых эфиров, претерпевающих затем
перегруппировку Кляйзена. Аналогичным образом еноляты сложных аллиловых эфиров
(образующиеся при действии литийизопроиилциклогексиламида на сложные эфиры)
перегруппировываются в γ,δ-ненасышенные кислоты.
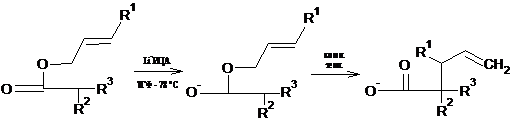
Простые диаллиловые эфиры претерпевают перегруппировку Кляйзена при
нагревании с трис (трифенилфосфнн) рутений(II) дихлоридом. Вероятно, последнее соединение катализирует
перегруппировку диаллилового эфира в аллилвиниловый эфир, который и
подвергается перегруппировке Кляйзена.
Известно много перегруппировок, которые можно считать аналогами
перегруппировки Кляйзена, например перегруппировки ArNHCH2CH=CH2, N-аллиленаминов R2C=CRNRCR2CR=CR2 и RC(OCH2CН=CH2)=NR.
8.6 Азо-перегруппировка Коупа
Превращение R1CH=N+R2CHR3CH2CH=CH2
в R3CH=N+R2-CHR1CH2CH=CH2
получило название азо-перегруппировки Коупа. Перегруппировки
пропаргилвиниловых эфиров приводят к алленовым альдегидам, кетонам, сложным
эфирам или амидам:

А= Н, R, OR, NR2
Аллилариловые тиоэфиры ArSCH2CH=СH2 не удается превратить в тиофенолы (тио-перегруппировка
Кляйзена) из-за неустойчивости последних; промежуточно образующиеся тиофенолы
реагируют далее с образованием бициклических соединений. Однако многие
аллилвинилсульфиды вступают в перегруппировку Кляйзена, что было использовано
для синтеза γ,β-ненасышенных альдегидов [19].
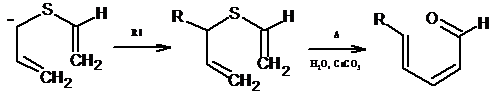
8.7 [2,3] - Сигматропная перегруппировка

Илиды серы, содержащие аллильную группу, при нагревании превращаются в
ненасыщенные сульфиды. Процесс представляет собой согласованную
[2,3]-сигматропную перегруппировку; показано, что аналогичным образом реагируют
илиды азота, сопряженные основания простых аллиловых эфиров, а также некоторые
другие системы. Перегруппировка была распространена даже на системы, целиком
состоящие из атомов углерода:
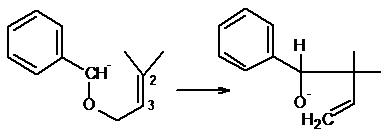
Поскольку реакция включает миграцию аллильной группы от серы, азота или
кислорода к соседнему отрицательно заряженному атому углерода, ее можно
рассматривать как особый случай перегруппировок Стивенса или Виттига. Однако в
указанных реакциях могут мигрировать и другие группы, а в данном случае
мигрирующей группой должна быть аллильная. При этом имеются две возможности: 1)
ион-радикальный механизм или механизм с участием ионной пары и 2) согласованная
перициклическая [2,3]-сигматропная перегруппировка. Эти два пути нетрудно
различить, так как последний всегда включает аллильный сдвиг (как в
перегруппировке Кляйзена), а первый - нет. Конечно, миграция групп, отличных от
аллильной, может происходить только по ион-радикальному механизму или по
механизму с участием ионной нары, поскольку согласованный механизм для
1.2-сдвига запрещен правилами орбитальной сим метрик.
Если группа OR или SR связана с отрицательно заряженным
атомом углерода, образующийся продукт легко гидролизуется. и в этом случае
реакция становится методом получения β,γ-ненасыщенных альдегидов [20]:
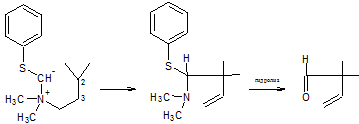
Список
литературы
) Робертс Дж., Касерио М. Основы органической химии. В
2-х т. Т.1. - М.: Мир, 1978. - 842 с. С. 519
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 269-270
) Робертс Дж., Касерио М. Основы органической химии. В
2-х т. Т.1. - М.: Мир, 1978. - 842 с. С. 514-516
) Реутов О.А., Курц А.Л., Бутин К.П. Органическая
химия. В 4-х частях. Ч.3. - М.:БИНОМ, Лаборатория знаний, 2004. - 544 с. С.
124-131
) Робертс Дж., Касерио М. Основы органической химии. В
2-х т. Т.1. - М.: Мир, 1978. - 842 с. С. 509-513
) Иванов В.Г., Горленко В.А, Гева О.Н.. Органическая
химия: Учеб. пособие для студ. высш. пед. учеб. заведений., М.: Мастерство,
2003. - 624 с. С. 138-139
) Агрономов А.Е. Избранные главы органической химии:
Учеб. пособие для вузов. - 2-е изд., М.: Химия, 1990. - 560 с. С. 53-54
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 270-272
) Сайкс П. Механизмы реакций в органической химии,
Изд. 3-е, - М.: Химия, - 1997, 320 с. С.232-233
10) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 201
11) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 192-193
) Курц А.Л., Брусова Г.П., Демьянович В.М. Одно- и
двухатомные спирты, простые эфиры и их сернистые аналоги. - Методическая
разработка для студентов кафедры органической химии, М.: МГУ им. М.В.
Ломоносова, 1999. - 65 с. С. 31-33
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 69
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 72
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 275-276
) Агрономов А.Е. Избранные главы органической химии:
Учеб. пособие для вузов. - 2-е изд., М.: Химия, 1990. - 560 с. С. 85
) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 90-93
) Марч Дж., Органическая химия. Реакции, механизмы и
структура. Углубленный курс для университетов и химических вузов: В 4-х т. Т.
4. Пер. с англ. - М.: Мир, 1988. - 468 с. С. 207-211
) Марч Дж., Органическая химия. Реакции, механизмы и
структура. Углубленный курс для университетов и химических вузов: В 4-х т. Т.
4. Пер. с англ. - М.: Мир, 1988. - 468 с. С. 213-214
) Курц А.Л., Брусова Г.П., Демьянович В.М. Одно- и
двухатомные спирты, простые эфиры и их сернистые аналоги. - Методическая разработка
для студентов кафедры органической химии, М.: МГУ им. М.В. Ломоносова, 1999. -
65 с. С. 23-26
) Курц А.Л., Брусова Г.П., Демьянович В.М. Одно- и
двухатомные спирты, простые эфиры и их сернистые аналоги. - Методическая
разработка для студентов кафедры органической химии, М.: МГУ им. М.В.
Ломоносова, 1999. - 65 с. С. 27-30
) Курц А.Л., Брусова Г.П., Демьянович В.М. Одно- и
двухатомные спирты, простые эфиры и их сернистые аналоги. - Методическая
разработка для студентов кафедры органической химии, М.: МГУ им. М.В.
Ломоносова, 1999. - 65 с. С. 30
) Робертс Дж., Касерио М. Основы органической химии. В
2-х т. Т.1. - М.: Мир, 1978. - 842 с. С. 519-522
25) Шабаров Ю.С. Органическая химия. - 4-е изд., М.:
Химия, 2002. - 847 с. С. 265-266