Факторы риска развития кардиоренального синдрома у больных системной красной волчанкой
МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ
УНИВЕРСИТЕТ
имени М.В. ЛОМОНОСОВА
Факультет фундаментальной медицины
ДИПЛОМНАЯ РАБОТА
Факторы риска развития
кардиоренального синдрома у больных системной красной волчанкой
Работа выполнена на кафедре
внутренних болезней ФФМ МГУ, на базе клиники нефрологии, внутренних и
профессиональных болезней им.Е.М. Тареева.
Автор:
Студентка 6 курса гр.604 Махмудова Салима Мурадовна
Руководители: Доцент кафедры внутренних болезней ФФМ МГУ,
к. м. н. Краснова Татьяна Николаевна
Ассистент кафедры внутренних болезней
ФФМ МГУБорисов Евгений Николаевич
Москва 2015
Список
сокращений
АГ - артериальная гипертензия
АД - артериальное давление
аКЛ - антитела к кардиолипину
АФС - антифосфолипидный синдром
ГП - гликопротеин
ДНК - дезоксирибонуклеиновая кислота
ЖКТ - желудочно-кишечный тракт
ИБС - ишемическая болезнь сердца
ИЛ - интерлейкин
ИМ - инфаркт миокарда
КИМ - комплекс интима-медиа
РЗ - ревматические заболевания
СКВ - системная красная волчанка
ССЗ - сердечно-сосудистое заболевание
ССС - сердечно-сосудистая система
ССО - сердечно-сосудистые осложнения
ТИА - транзиторные ишемические атаки
ТЭЛА - тромбоэмболия легочной артерии
УЗДГ - ультразвуковая допплерография
УЗИ - ультразвуковое исследование
ФНО - фактор некроза опухоли
ЦИК - циркулирующие иммунные комплексы
ЭКГ - электрокардиограмма
ЭхоКГ - электрокардиография
eNos - эндотелиальная NO-синтаза- иммуноглобулины
класса G- иммуноглобулины класса M- мутантная аллель-
метилентетрогидрофолатредуктаза- никотинамидадениндинуклеотидфосфат- тканевой
фактор моноцитов- тромбоксан
Содержание
Введение
Обзор литературы
Основные механизмы развития сердечно-сосудистых заболеваний при
системной красной волчанке и антифосфолипидном синдроме
Антифосфолипидный синдром: поражение ССС и почек, тромботические
осложнения
Роль тромбофилии в развитии тромботических осложнений
Полиморфизм генов тромбофилии
Кардиоренальный синдром
Основные патофизиологические механизмы формирования
кардиоренального синдрома
Материалы и методы
Результаты исследования
Обсуждение результатов исследования
Выводы
Список литературы
Введение
Системная красная волчанка (СКВ) - системное аутоиммунное
ревматическое заболевание неизвестной этиологии, характеризующееся
гиперпродукцией широкого спектра органонеспецифических аутоантител к различным
компонентам ядра клетки и иммунных комплексов, вызывающих иммуновоспалительное
повреждение внутренних органов. Болезнь наиболее часто развивается у женщин
репродуктивного возраста: риск обострения СКВ возрастает во время беременности
и в послеродовом периоде.
Системная красная волчанка, по данным различных авторов,
встречается с частотой 2,7-4,8 на 100 000 населения, соотношение больных женщин
и мужчин от 8: 1 до 10: 1 [3]. Пик заболеваемости приходится на возраст 15-25
лет. Смертность при СКВ в 3 раза выше, чем в популяции [4].
Антифосфолипидный синдром (АФС) - симптомокомплекс,
характеризующийся венозными и/или артериальными тромбозами, акушерской
патологией (невынашивание в I и II триместрах беременности, преждевременные роды),
реже тромбоцитопенией, а также другими (сердечно-сосудистыми, неврологическими,
кожными т.д.) проявлениями связанный с гиперпродукцией антител к кардиолипину
(аКЛ). АФС может развиваться у 20-30% с СКВ [11].
Стоит отметить, что этиология и патогенез СКВ и АФС во многих
аспектах не изучены или изучены недостаточно.
АФС относится к числу наиболее актуальных
мультидисциплинарных проблем современной медицины и может рассматриваться как
уникальная модель аутоиммунной тромботической васкулопатии. Изучение
антифосфолипидного синдрома имеет существенное значение для понимания
взаимоотношений между такими фундаментальными патологическими процессами,
составляющими основу сосудистой патологии при заболеваниях человека, как
воспаление, атеросклероз и гиперкоагуляция.
красная волчанка кардиоренальный синдром
Нарушения в работе свертывающей и противосвертывающей
системах крови не могут быть объяснены только прямым воздействием аКЛ, поиск
дополнительных факторов развития осложнений в рамках АФС необходим для более
полного представления о патогенезе заболевания, а значит и для более
эффективного лечения. В связи с увеличением доступности методов генетической
диагностики, все актуальнее становится определение вклада наследственных
факторов в вероятность развития и характер течения АФС.
Проявления СКВ чрезвычайно разнообразны, что определяется
множественностью поражения органов и систем, характером течения, фазой и
степенью активности воспалительного процесса. Поражение сердечно-сосудистой
системы наблюдается на различных этапах течения болезни [11].
Среди причин поражения ССС рассматриваются васкулит и/или
невоспалительная васкулопатия (обусловленная АФС).
Наиболее часто встречаются перикардиты, имеющие тенденцию к
рецидивам. Значительно чаще, чем это представлялось прежде, поражается эндокард
в виде развития бородавчатого эндокардита (волчаночный эндокардит) на створках
митрального, а также аортального или трехстворчатого клапанов. При длительном
течении процесса можно выявить признаки недостаточности соответствующего
клапана (признаков стенозирования отверстия, как правило, не отмечается).
Поражение сосудов может проявляться в виде синдрома Рейно
и/или капилляритами: приступообразно развивающиеся расстройства артериального
кровоснабжения кистей и/или стоп, возникающие под воздействием холода или волнений.
Во время обострения отмечаются парестезии, кожа пальцев становится бледной
и/или цианотичной, пальцы холодные. [23]. Кроме этого отмечается высокая
частота тромбоэмболических состояний, а также микроангиопатии.
Морфологически тромботическая микроангиопатия характеризуется
образованием множественных фибриновых тромбов в капиллярах, артериолах и
артериях, которые в сочетании с артерио - и артериолосклерозом, фиброзом
сосудистой стенки приводят к окклюзии сосудов, склерозу стромы, нарушениям
микроциркуляции и ишемии тканей и органов (нередко почек).
Клиническими проявлениями тромботической микроангиопатии при
АФС являются артериальная гипертензия разной степени выраженности, нарушение
функции почек и умеренный мочевой синдром, чаще всего представленный изолированной
протеинурией.
В литературе широко обсуждается раннее развитие атеросклероза
при аутоиммуных заболеваниях.
Известно, что у пациентов с системной красной волчанкой риск
возникновения тромбоза выше, чем в популяции. По данным 21 исследования, включавших
1428 больных СКВ и волчаночно-подобными синдромами, тромботические осложнения
были отмечены у 24% пациентов [1]. Иммунокомплексное воспаление, а более точно
- "провоспалительные" цитокины, характерные для СКВ, могут
способствовать прокоагулянтным нарушениям в системе гемостаза, приводящим к
тромбозу [2, 3, 4, 5, 6]. Эти нарушения, а именно - активация свертывающих
факторов, синтеза тромбина и фибрина и угнетение фибринолиза и имеют место при
СКВ [1, 7, 8, 9, 10, 11]. Помимо воспаления при СКВ существуют и другие факторы
риска тромбообразования, в том числе - антифосфолипидные антитела (аФЛ),
мутации генов некоторых факторов свертывания крови, гипергомоцистеинемия,
раннее атеросклеротическое повреждение сосудов, которые необходимо учитывать
при выборе тактики лечения и анализе исходов.
Согласно современным международным статистическим данным,
поражение почек развивается у примерно 50% больных системной красной волчанкой,
что увеличивает риск развития почечной недостаточности, сердечно-сосудистой
патологии и смерти. Уже на момент постановки диагноза около 35% взрослых,
страдающих СКВ, имеют те или иные клинические признаки нефрита; у 50-60%
пациентов люпус-нефрит (волчаночный нефрит) развивается в течение первых 10 лет
болезни. [27]
Поражения сердца и почек широко распространены в популяции и
часто сосуществуют, повышая смертность и риск осложнений. Более того, на
сегодняшний день имеются веские основания обсуждать общность патогенеза,
факторов прогрессирования хронической болезни почек и хронической болезни
сердца, необходимость особых подходов к их комбинированному лечению.
Двусторонне направленные взаимоотношения сердце - почки, при котором
патофизиологическое нарушение в одном из них может приводить к дисфункции
другого, определено понятием "кардиоренальный синдром" (КРС) [5,53].
Поражение почек при СКВ является одним из наиболее серьезных
висцеритов, зачастую определяющим тяжесть заболевания и приводящим к ранней
инвалидизации больных (Насонова В.А., Насонов Е.Л., 2007). Даже при
использовании современных иммуносупрессивных препаратов 20-летняя выживаемость
не превышает 20% (Захарова Е.В. и др., 2003; Соловьев С.К., 2008). Клинические
проявления волчаночной нефропатии встречаются в 50-70% всех случаев СКВ, тогда
как морфологические признаки нефропатии обнаруживаются у 100% больных (Захарова
Е.В. и др., 2003; Cameron J. S., 1999). В настоящее время убедительно доказано, что СКВ -
классическое проявление 5 типа кардиоренального синдрома, когда поражение
сердца и почек развивается одновременно. [83].
Снижение продолжительности жизни у
пациентов с системными заболеваниями соединительной ткани, несмотря на
совершенствование методов диагностики и лечения, остается актуальной проблемой.
Кардиоваскулярные осложнения, обусловленные развитием артериальной гипертензии
и ранним атеросклеротическим поражением сосудов, являются одними из основных
причин летальности при ревматических заболеваниях.
Учитывая все вышесказанное, представляется актуальным изучить
особенности сочетанного поражения сердечно-сосудистой системы и почек у больных
СКВ.
Цель: выявить факторы риска развития кардиоренального
синдрома у больных системной красной волчанкой
Для достижения цели были поставлены следующие задачи:
· Определить частоту кардиоренального
синдрома (КРС) при СКВ
· Оценить вклад антифосфолипидного синдрома
в развитие КРС у больных СКВ
· Выявить особенности сердечно-сосудистой
патологии при волчаночном нефрите (ВН)
· Оценить роль полиморфизма генов
тромбофилии в развитии кардиоренального синдрома у больных СКВ
Обзор
литературы
Поражение сердечно-сосудистой системы с ранним развитием
атеросклероза характерно для ревматических заболеваний (РЗ).
Среди причин смерти при различных формах РЗ преобладает
сердечно-сосудистая патология, а именно сосудистые катастрофы типа инфаркта
миокарда и инсульта, связанные прежде всего с ускоренным развитием
атеросклероза [20,21,22,23]. Поражения сердца являются одной из важных причин
тяжести состояния и смертности пациентов с СКВ [23-25]. Так, при аутопсии 106
больных СКВ, по данным C. Bastos и соавт. [26], кардиоваскулярные нарушения
отсутствовали лишь у 7,5 % пациентов. Сосудистые катастрофы относятся к частым
причинам преждевременной смерти больных СКВ и обусловлены прежде всего
атеросклеротическим поражением сосудов и тромботическими осложнениями [19, 22,
27-29]. В течение 10-15 лет от начала болезни кардиоваскулярные осложнения
развиваются более чем у 1/3 пациентов и у многих сразу приводят к летальному
исходу. Кардиоваскулярная патология является причиной летальных исходов более
чем у половины больных СКВ [12, 31]. У больных СКВ отмечено двукратное
увеличение смертности от инфаркта миокарда и инсульта [32, 33]. При этом ускоренное
развитие атеросклероза служит своеобразным внесуставным (системным) проявлением
СКВ.
При РЗ актуальна также проблема кардиоваскулярной
безопасности в связи с использованием нестероидных противовоспалительных
препаратов. При этих заболеваниях системный воспалительный процесс
ассоциируется с увеличением риска сосудистых катастроф независимо от
классических факторов риска атеротромбоза [34].
РЗ относятся к процессам, в развитии которых ведущую роль
играют аутоиммунные механизмы [35-39]. В патогенезе РЗ большое внимание
уделяется нарушению функции эндотелия, который не только имеет большое значение
в системе гемостаза за счет своих антитромбогенных свойств, но и является
важным модулятором многих физиологических свойств сосудистой стенки [40, 41].
Имеются доказательства участия эндотелиальной дисфункции в атерогенезе, ишемии
миокарда, возникновении коронарного тромбоза, ремоделирования левого желудочка
и сердечной недостаточности [42-44]. В то же время выявлена прямая зависимость
между дисфункцией эндотелия и риском сердечно-сосудистых осложнений, что
объясняется непосредственным участием оксида азота в клеточных реакциях,
контроле тромбоцитарно-сосудистых взаимоотношений и сосудистого тонуса [45,
46]. Накапливается все больше фактов, указывающих на значение воспалительного
процесса в сосудистой стенке как фактора формирования атеросклероза и
возможного возникновения тромбоза [47, 48]. С этим связано раннее и частое
развитие атеросклеротических изменений у больных ревматического профиля.
Регуляция сосудистых и клеточных воспалительных реакций осуществляется большим
числом биологически активных веществ, среди которых особое место занимают
цитокины - низкомолекулярные белковые молекулы, обеспечивающие процессы
межклеточных коммуникаций [35, 36, 49, 50]. Утверждение о роли цитокинов в
повреждении эндотелия при атеросклерозе нашло подтверждение при изучении таких
медиаторов воспаления, как интерлейкин-1, интерлейкин-3, фосфолипаза А2,
интегрин МАС-1, при этом процессе. Уровень ФНО-a коррелирует с выраженностью атеросклеротического
поражения сонных артерий и эндотелиальной дисфункцией при СКВ [37]. Получены
данные о корреляции ФНО-a с фракцией выброса левого желудочка при хронической
сердечной недостаточности, что обусловлено хронической гипоксией и эндотоксемией
[28]. По мере прогрессирования этого заболевания под воздействием возрастающей
цитокиновой и нейрогуморальной активации определяется прогрессирующее
повреждение эндотелия, приводящее к нарушению выработки оксида азота и других
вазодилататоров эндотелиального происхождения. Иммунологическими маркерами
сосудистых поражений при РЗ являются антинейтрофильные цитоплазматические
антитела, которые определяются у 47% больных РЗ, антикардиолипиновые антитела,
определяющиеся почти у половины больных СКВ и васкулитами и циркулирующие
иммунные комплексы (ЦИК), которые являются не только маркерами РЗ, но и
патогенетическими факторами [19]. В патогенезе РЗ определенную роль могут
играть нарушения липидного обмена или так называемые дислипопротеидемии,
поскольку конъюгированный холестерин и его аналоги обладают высокоиммуногенными
свойствами. Иммунные реакции с участием антител возникают в случае, когда в
качестве аутоантигена выступают производные холестерина - эстерифицированный
холестерин и стероидные гормоны [10]. При обследовании больных с СКВ
установлено, что уровень холестерина в преципитатах иммунных комплексов был
значительно повышен, что позволяет обсуждать роль холестерина иммунных
комплексов в атерогенезе у больных СКВ [11]. В то же время половые стероиды у
женщин, прежде всего эстрогены, не только предрасполагают к возникновению
аутоиммунных болезней [22, 43], но и оказывают протективное действие в
отношении раннего развития атерогенных дислипопротеидемий - ведущего фактора
возникновения атеросклероза [14, 26].З.С. Алекберова и соавт. [35] у 40 женщин
с СКВ исследовали атерогенность сыворотки крови в культуре мышиных макрофагов
по методу В.В. Тертова и соавт. [45]. Выявлена прямая зависимость атерогенности
сыворотки от активности заболевания, что указывает на роль иммунологических
нарушений в атерогенезе у больных СКВ. Ассоциации с показателями липидного
спектра крови не было отмечено.
Поражение сердечно-сосудистой системы при РЗ имеет место
практически при всех нозологических формах диффузных заболеваний соединительной
ткани и системных васкулитах, определяя течение этих прогностически
неблагоприятных заболеваний и качество жизни больных [11, 13, 54]. По данным
В.А. Якушевой и В.И. Мазурова [52], среди больных СКВ с ИБС более выраженные
ишемические изменения были обнаружены в группе с системными проявлениями и
продолжительностью заболевания более 5 лет. Авторы считают, что хронический
воспалительный процесс у больных СКВ является ключевым моментом в развитии
эндотелиальной дисфункции, имеющей значение при формировании атеросклероза.
Кардиальная патология, по данным эхокардиографии, была выявлена у 45,8% больных
СКВ, причем наиболее часто встречались пролапс митрального клапана и расширение
желудочков сердца [7]. При обследовании 75 больных СКВ диастолическая дисфункция
установлена у 41,3%, особенно при более позднем назначении базисной терапии
антиревматическими препаратами [71]. По данным И.В. Иткиной [12], при
обследовании 60 женщин, больных СКВ, поражения сердца проявлялись нарушением
ритма, адгезивным перикардитом и снижением сократительной способности миокарда.
По данным других авторов [3, 4], поражения сердца при СКВ характеризовались
явлениями экссудативного перикардита, миокардита, пролапсом митрального
клапана. При проведении холтеровского мониторирования ЭКГ у 61,5% больных СКВ
наблюдались ишемические изменения, причем особенностью течения стенокардии
можно считать преобладание безболевых эпизодов у 70,8% больных [5]. Изменения
функционального состояния миокарда, коррелирующие с активностью заболевания,
были выявлены у 47 больных СКВ без клинических признаков поражения сердца [6].
Частота поражений сердца при СКВ, по данным разных
исследователей, колеблется от 54 до 73,3%. Наиболее часто регистрируется
перикардит, характеризующийся преимущественно мягким течением, затем -
поражение клапанного аппарата, в основном митрального клапана, и поражение
миокарда. Имеют место также нарушения функции левого желудочка, его
гипертрофия, наличие систолической сердечной недостаточности и легочной
гипертензии [17-21]. При эхокардиографическом исследовании [12] у 108 больных
СКВ, поражение сердца было выявлено у всех пациентов, имеющих клиническую
активность. В ряде исследований отмечена взаимосвязь между повышением
активности процесса и присоединением сердечно-сосудистых осложнений [43, 44].
Т.В. Попкова и соавт. [15], определяя индивидуальный суммарный риск коронарных
осложнений при СКВ, установили, что он является высоким. Это было обусловлено
изменениями в спектре атерогенных липидов и липопротеидов. Развитие аритмий
может быть вторичным вследствие миокардита, перикардита или ишемии в результате
коронарного васкулита. Развитие мерцания предсердий встречается нечасто.
Имеется несколько сообщений о возникновении фибрилляции предсердий после начала
пульс-терапии метилпреднизолоном [16,17]. У больных СКВ в отсутствие изменений
центральной гемодинамики выявлены значительные нарушения диастолической
функции, в основном правого желудочка, а также легочного кровотока. При
исследовании липидного обмена констатировано повышение атерогенности сыворотки
крови [28]. При обследовании больных СКВ с применением перфузионной
томосцинтиграфии миокарда в покое и в сочетании с пробой с физической нагрузкой
на велоэргометре были выявлены нарушения перфузии миокарда различного
происхождения, в том числе характерные для рубцового повреждения,
мелкоочагового фиброза и преходящей ишемии миокарда различного генеза:
вследствие нарушений микроциркуляции и атеросклеротического поражения
магистральных артерий [38]. Ю.В. Пак и Н.Н. Бажанов [40] получили данные о
крайне редком поражении основных стволов коронарных артерий при СКВ. Авторы
считают, что имеющаяся достоверная ишемия миокарда у больных СКВ с вторичным
антифосфолипидным синдромом в возрасте 15-35 лет может быть следствием
нарушения перфузии миокарда на микроциркуляторном уровне за счет воспаления или
микротромбоза сосудов миокарда.
У пациентов с СКВ часто имеется латентное поражение сердца.
Иммунологические исследования показали экспрессию HLA II класса антигенов к
кардиальным фибробластам [31]. При обследовании 75 женщин и 6 мужчин с СКВ была
выявлена диастолическая дисфункция сердца. При нарастании степени синдрома
Рейно ухудшалась диастолическая функция обоих желудочков сердца, что не
исключает распространенного характера микроциркуляторных изменений при СКВ
[22]. Изменения сердца, обнаруженные у 66,7% больных СКВ, выражались в
уплотнении створок и хорд в клапанном аппарате с преимущественным поражением
митрального клапана, поражении сердца в результате легочной гипертензии с
формированием хронического легочного сердца. По данным A. Kane и соавт. [23],
поражение сердца имело место у 48,3% больных. Нарушения ритма и проводимости
были выявлены у 26,6% пациентов. При холтеровском мониторировании ЭКГ и
эхокардиографии отмечена ранняя дисфункция кардиоваскулярной системы у
пациентов с СКВ, не имеющих явных кардиальных симптомов [58], уменьшение
наполнения правого желудочка найдено у значительного числа больных СКВ,
включенных в исследование. Эти изменения коррелировали с систолическим легочным
давлением и нарушением наполнения левого желудочка. В группе больных СКВ индекс
массы левого желудочка был значительно выше, чем в контроле, а фракция выброса
- ниже, чем в других группах. Анализ региональных фракций выброса показал
значительные нарушения проксимального сегмента внутрижелудочковой перегородки.
У пациентов с СКВ в основном отмечалась гипертрофия левого желудочка со
снижением фракции выброса. По данным А.В. Волкова и соавт. [50], при
обследовании 100 больных СКВ были выявлены нарушения ритма и проводимости,
дилатация левого предсердия, дилатация и гипертрофия левого желудочка,
нарушение диастолического расслабления миокарда желудочков, снижение
сократительной способности миокарда.
Потеря иммунной толерантности к
собственным, в первую очередь ядерным, антигенам приводит к продукции множества
комплемент-связывающих аутоантител к компонентам клеточного ядра, цитоплазмы и
мембран, в частности к двухцепочечной ДНК и нуклеосомам. Аутоантитела оказывают
как прямое повреждающее действие на различные органы и ткани, так и
опосредованное - через формирование иммунных комплексов и активацию системы
комплемента.
Волчаночный нефрит (ВН) представляет собой
одно из наиболее серьезных и прогностически значимых проявлений СКВ. Механизм
развития люпус-нефрита иммунокомплексный. Связывание антител к ДНК и других
аутоантител с базальной мембраной почечных клубочков ведет к активации
комплемента и рекрутированию клеток воспаления в гломерулы.
Характерно также не только иммунокомплексное, но и
тромботическое поражение сосудов, последнее обусловлено наличием антител к
кардиолипину, а также развитием АФС и вторичного ДВС. Таким образом, системные
повреждения имеют смешанный (цитотоксический, иммунокомплексный и
тромботический) генез.
Хроническое аутоиммунное воспаление
приводит к повреждению сосудистого эндотелия и нарушению его функции, которое
может выявляться уже на ранней стадии заболевания [А.П. Ребров и соавт., 2008;
Н.Л. Козловская и соавт., 2010; M. Frieri, 2012; A. Y. Funakubo, 2012].
Возникающий дисбаланс между эндотелий-зависимыми вазодилятирующими и
вазоконстрикторными факторами, в сторону увеличения выработки последних,
инициирует пролиферацию гладкомышечных клеток сосудов. В результате изменяется
соотношение толщины комплекса интима-медиа, увеличиваются показатели ригидности
артерий. Нарушение сосудистой архитектоники значительно ускоряет процесс
развития атеросклероза и является фактором риска сердечно-сосудистых осложнений
[Д.С. Новикова и соавт., 2009; В.И. Маколкин, 2011; Ю.А. Карпов, 2012; C. W.
Chin et al., 2013]. Параметры жесткости артерий у больных ревматического
профиля представляет интерес как предиктор развития кардиоваскулярной
патологии.
Основные
механизмы развития сердечно-сосудистых заболеваний при системной красной
волчанке и антифосфолипидном синдроме
Более глубокое понимание механизмов, способствующих
повреждению сосудов, позволило оценить роль провоспалительных путей в развитии
и распространении атеросклероза. Некоторые механизмы, приводящие к образованию
атеросклеротической бляшки и, следовательно, сердечно-сосудистому заболеванию,
могут иметь место при наличии аутоиммунного поражения, однако каждое
заболевание обладает индивидуальными иммунологическими особенностями, которые
обусловливают развитие специфических проатерогенных путей [5-7, 16, 24, 61-68].
Атеросклероз характеризуется накоплением липидных частиц, иммунных клеток,
аутоантител, аутоантигенов, продукцией воспалительных цитокинов. Все эти
компоненты приводят к постепенному утолщению слоя интимы, таким образом,
способствуя уменьшению эластичности, сужению просвета артерии, снижению
кровотока, разрыву бляшки и, наконец, сердечно-сосудистому событию [69,70].
При аутоиммунном поражении появляются определенные факторы
риска, которые способствуют более раннему повреждению сосудов. Нарушенная
функция эндотелия и увеличение толщины комплекса интима-медиа ускоряют развитие
атеросклероза. Данные изменения более выражены при полиаутоиммунном поражении
(при СКВ и АФС), когда факторы риска суммируются и атеросклероз развивается
"раньше".
При аутоиммуных ревматических заболеваниях "раннее"
развитие атеросклероза обусловлено накоплением традиционных факторов риска
(ТФР) сердечно-сосудистых заболеваний; аутоиммунным воспалением, составляющим
основу патогенеза этих заболеваний; побочными эффектами противоревматической
терапии (van Doorum S. и соавт., 2002; Насонов Е.Л., 2005, Gazi I. F., 2007;
Westerwell P. E. и соавт., 2007).
В ряде исследований подчеркивается важная роль ТФР (возраст,
артериальная гипертензия, дислипидемия, сахарный диабет, курение, избыточный вес,
малоподвижный образ жизни, отягощенная наследственность по ССЗ, менопауза и
др.) в развитии атеросклероза у пациентов с СКВ (Gladman D. D. и соавт, 1987;
Manzi S. и соавт., 1999; Svenungsson E. и соавт., 2001; Gazi I. F., 2007).
Авторы ряда работ ранние проявления атеросклеротического
поражения сосудов связывают с активностью и длительностью СКВ, тяжестью
повреждения внутренних органов (Font J. и соавт., 2002), нефритом (Bono L. И
соавт., 1999; Falaschi F. и соавт., 2001), кумулятивной дозой глюкокортикоидов
(Manzi S. И соавт., 1997; Bruce I. N. и соавт., 2003).
При хронической активации иммунного ответа создаются условия
для гиперпродукции "провоспалительных" цитокинов и относительной
недостаточности синтеза "антивоспалительных" медиаторов (Hansson G. K. и соавт., 2002; Von der Thusen J. H. и соавт., 2003).
Результатом этого дисбаланса является развитие тех сосудистых нарушений,
которые составляют основу атеротромбоза: дисфункция эндотелия, вазоконстрикция,
перекисное окисление липидов и липопротеидов, гиперкоагуляция. Многие
иммунонологические маркеры атерослероза (белки острой фазы воспаления,
"провоспалительные" цитокины, клеточные молекулы адгезии,
аутоантитела, иммунные комплексы), с одной стороны, являются
"предикторами" атеротромботических осложнений и ассоциируются с ТФР
ССЗ, с другой - отражают течение хронического воспалительного процесса при СКВ
(Szmitko P. E. и соавт., 2003; Насонов Е.Л., 2004).
Антифосфолипидный
синдром: поражение ССС и почек, тромботические осложнения
В 1975 г. G. Hughes описал синдром, названный впоследствии
антифосфолипидным, основными признаками которого являются артериальные и
венозные тромбозы, а также повышение уровня антикардиолипиновых антител - IgG-,
IgM-аКЛ. По современным представлениям, основу антифосфолипидного синдрома
составляет своеобразная васкулопатия, обусловленная невоспалительным или
тромботическим поражением сосудов и заканчивающаяся их окклюзией [27].
Кардиологические клинические проявления антифосфолипидного синдрома,
ассоциирующиеся с синтезом антикардиолипиновых антител, связаны с локализацией
процесса на клапанах сердца, в процесс вовлекаются также коронарные артерии,
сосуды дуги аорты, сонные артерии, сосуды легких и мелкие кожные сосуды [33,
44]. При высоком титре антител к кардиолипинам наиболее часто регистрируются
поражения сердечно-сосудистой системы. Чувствительность и специфичность
показателя высокого уровня антикардиолипинов являлись предиктором сердечных
нарушений в 78 и 74% соответственно [45]. Сочетание антифосфолипидных антител с
повреждением миокарда указывает на то, что эти антитела могут играть
значительную роль в патогенетических механизмах поражения клапанов сердца при
СКВ [10, 17]. Отмеченная взаимосвязь между увеличением титра антикардиолипинов
и выраженностью систолической дисфункции левого желудочка предполагает
возможную патогенетическую роль антифосфолипидных антител в формировании
гипокинезии миокарда у больных СКВ [9, 18, 19]. В настоящее время обсуждается
взаимосвязь между продукцией антифосфолипидных антител и ускоренным развитием
атеросклероза. Однако даже при тяжелом атеросклерозе, приводящем к полной
окклюзии просвета сосуда, как правило, отсутствуют другие клинические
проявления, характерные для антифосфолипидного синдрома. Существует немало
фактов, свидетельствующих о взаимосвязи между аутоиммунными нарушениями и
дислипопротеинемиями. Так, при изучении больных СКВ и экспериментальной модели
этой патологии - мышей линии NZB/NZW - было показано, что в ряде случаев
нарушения липидного обмена выявляются уже на ранней стадии заболевания. Частота
тромбозов была выше в случаях, когда гиперпродукция антифосфолипидных антител
сочеталась с нарушениями липидного обмена в виде триглицеридемии [10]. Для
ранней оценки атеросклеротического поражения сосудов в качестве скринингового,
доступного и относительно недорогого метода все шире используется
ультразвуковое исследование (УЗИ) сонных артерий, предложено также определение
толщины комплекса интима-медиа (КИМ) при УЗИ сонных артерий. В многочисленных
исследованиях выявлена взаимосвязь этого показателя с риском развития
кардиальных и цереброваскулярных осложнений, особенно отчетливая при наличии
артериальной гипертензии [9, 11]. Многие авторы расценивают утолщение КИМ
сонной артерии как независимый фактор риска сердечно-сосудистых осложнений и
предиктор ИБС. Установлена взаимосвязь между уровнем систолического АД и
толщиной КИМ общей сонной артерии. Комбинированный показатель "толщина КИМ
+ бляшка" тесно коррелировал с возрастом больных, мужским полом,
гипергликемией [12]. В международном многоцентровом двойном слепом
рандомизированном исследовании ELSA выявлены наиболее сильные взаимосвязи между
показателями УЗИ сонных артерий и возрастом, систолическим и пульсовым АД.
Значения толщины КИМ коррелировали с возрастом, среднесуточным пульсовым АД,
уровнем холестерина липопротеидов низкой плотности, мужским полом, уровнем
триглицеридов, курением и систолическим АД. При проведении ультрасонографии
общих сонных артерий у 20 больных СКВ атеросклеротические бляшки обнаружены у
30%, определение толщины КИМ расценивается как наиболее ценный диагностический
метод оценки состояния периферических артерий. Дуплексное сканирование общей
сонной артерии было выполнено у 100 больных СКВ. Выявлены статистически
значимое увеличение ее диаметра, а также толщины КИМ (0,99-0,08 и 0,84-0,04 мм;
p<0,05) и уменьшение максимальной скорости кровотока у пациентов, у которых
заболевание начало развиваться в возрасте старше 50 лет [13]. Однако при
исследовании сосудов конечностей подобных различий не было обнаружено, так что
у авторов вызвало сомнение влияние атеросклеротических процессов на развитие
ишемических нарушений. Этот факт подтверждается и другими исследователями,
которые не нашли атеросклеротических изменений артерий среднего калибра у
больных СКВ с тяжелыми дигитальными гангренами [4, 5]. Сообщения о выраженности
систолической дисфункции левого желудочка у больных СКВ довольно противоречивы
[10-11]. Так, по данным одних исследователей, не было отмечено снижения
систолической функции при наличии значительного снижения диастолической функции
левого желудочка [8]. Средние значения фракции выброса левого желудочка (LVEF)
при СКВ составляли 59%, при СКВ - 61%. Уменьшение LVEF (менее 50%) было
установлено у 19% пациентов СКВ и у 8% - СКВ [19]. Однако, по данным других исследователей,
у больных СКВ наряду с диастолическими отмечались нарушения систолической
функции левого желудочка, которые сопровождались развитием застойной сердечной
недостаточности и особенно проявлялись при физической нагрузке [12, 20]. При
обследовании 75 больных СКВ диастолическая дисфункция была выявлена у 41,3%,
особенно при наличии ревматоидного фактора, с быстропрогрессирующим течением
СКВ, более поздним назначением базисной терапии модифицирующими болезнь
антиревматическими препаратами [22].
У 9% больных СКВ наблюдается, как минимум, один эпизод
тромботических осложнений в течение первых 10 лет от начала заболевания [60].
Около 26% случаев летального исхода у больных с СКВ связаны с тромбозами [4].
АФС значительно повышает вероятность развития тромботических осложнений (в 5 -
16 раз) у пациентов с СКВ [18]. Появление аКЛ увеличивает риск тромботических
осложнений в 1,6 раз при любом титре и в 3,2 раза при высоких титрах (более 10
МЕ/мл для аКЛ IgG и более 7 МЕ/мл для аКЛ IgM). [21] У пациентов в возрасте до 50 лет развитие
АФС с повышением уровня аКЛ является отдельным значимым фактором риска
проявлений микротромбозов, артериальных и венозных тромбозов. У людей старшего
возраста связь между АФС и тромботическими осложнениями менее выражена, ввиду
значительного влияния других факторов (болезни сердечнососудистой системы,
сахарный диабет, вредные привычки, образ жизни) [19]. Характерной особенностью
АФС является высокий риск рецидивов венозных и артериальных тромбозов. При этом
у больных с первым тромбозом в артериальном русле повторные тромбозы также чаще
развиваются в артериях. Если же первым тромбозом был венозный, то повторные
тромбозы, как правило, происходят в венозном русле. [3] У пациентов с высоким
уровнем аКЛ IgG в сыворотке повторные нарушения мозгового кровообращения
развивались в более ранние сроки, чем у пациентов с низким уровнем аКЛ. [51]
Венозный тромбоз, особенно тромбоз глубоких вен нижних
конечностей - частое проявление АФС, в том числе в дебюте заболевания. В общей
популяции аКЛ выявляются примерно у 10% пациентов с венозными тромбозами. [21.
При АФС частота венозных тромбозов колеблется от 29 до 55%. Тромбы обычно
локализуются в глубоких венах нижних конечностей, но нередко - в печеночных,
портальных, поверхностных венах и др. Тромбоз центральной вены надпочечников
может приводить к надпочечниковой недостаточности. [3] Наиболее опасным
осложнением венозных тромбозов является тромбоэмболия легочной артерии (ТЭЛА).
Летальность от ТЭЛА без лечения составляет 30%. В случае нелетальной ТЭЛА наблюдаются
инфаркт легкого, хроническая тромбоэмболическая легочная гипертензия,
правожелудочковая сердечная недостаточность. [1] Появление сердечной
недостаточности говорит о запущенности заболевания и снижает эффективность
лечения. Такие больные умирают в течение ближайших 5-6 лет. [2]
Частота артериальных тромбозов в рамках АФС составляет от 20%
до 34%. [3] Они проявляются ишемией и/или инфарктами мозга, коронарных артерий,
нарушениями периферического кровообращения. Наиболее частой локализацией
тромбоза (в 50% случаев) являются мозговые, реже коронарные артерии. [12] К
редким проявлениям АФС относится тромбоз крупных артерий, а также восходящего
отдела аорты (с развитием синдрома дуги аорты) и брюшной аорты. Инсульт и
транзиторные ишемические атаки (ТИА) - частые клинические проявления АФС.
Ведущей причиной поражения ЦНС является тромбоз мозговых артерий, который
приводит к ишемии мозга. [19] Тромбоз коронарных артерий является одним из
возможных проявлений артериального тромбоза у больных с аКЛ в крови. Инфаркт
миокарда (ИМ) развивается приблизительно у 5% аКЛ положительных больных. [61] В
целом тромботические осложнения в артериальном русле могут вызывать поражение
почек, печени, легких, сетчатки, костей, желудочно-кишечного тракта (ЖКТ).
Одной из важных составляющих клинических проявлений АФС
являются ишемические поражения внутренних органов. Наиболее часто
недостаточность кровообращения в следствии микроангиопатии наблюдается в
почках, надпочечниках, сердце, головном мозге и сетчатке. Также ишемические
поражения приводят к нарушениям функций поджелудочной железы, печени,
кишечника. [4] Наличие системной микротромботической патологии ухудшает прогноз
течения СКВ у больных с АФС, повышает их летальность. [3]
Почечная патология при АФС весьма разнообразна. У большинства
пациентов наблюдается бессимптомная умеренная протеинурия (менее 2 г в сут),
без нарушения функции почек, но может развиваться острая почечная
недостаточность с выраженной протеинурией (вплоть до нефротического синдрома),
активным мочевым осадком и артериальной гипертонией. Поражение почек при АФС
связано с внутриклубочковым микротромбозом и определяется как "почечная
тромботическая микроангиопатия".
Частым осложнением АФС является артериальная гипертензия,
которая может быть лабильной, нередко ассоциирующейся с сетчатым ливедо и
поражением церебральных артерий в рамках синдрома Снеддона (артериальная
гипертензия, рецидивирующие тромбозы мозговых артерий и мраморный рисунок
кожи), или стабильной, злокачественной, проявляющейся симптомами гипертонической
энцефалопатии. Развитие артериальной гипертензии при АФС может быть связано со
многими причинами, в том числе с тромбозом почечных сосудов, инфарктом почек,
тромбозом брюшного отдела аорты ("псевдокоарктация") и
интрагломерулярным тромбозом почек. Отмечена связь между гиперпродукцией
антифосфолипидных антител и развитием фибромышечной дисплазии почечных артерий.
Роль
тромбофилии в развитии тромботических осложнений
Начало поисков наследуемой тромбофилии было положено работами
О. Эгеберга. В 1965 г. он описал норвежскую семью, в которой склонность к
венозным тромбозам наблюдалась на протяжении нескольких поколений.
Тромботические явления были отмечены в молодом возрасте. Изучение крови больных
позволило обнаружить выраженное уменьшение (на 40 - 50%) антитромбина III.
Более поздние исследования Г. Шаш показали возможность различных форм дефекта
антитромбина III, который встречается довольно часто. У больных с повторными
тромбозами его выявляли у 5 - 7%, при анализе лиц с однократно диагностированным
венозным тромбозом он встречался в 1% случаев. Среди здоровых лиц его выявляют
у 1 на 5000 обследованных.
Наледственные тромбофилии изучаются достаточно интенсивно во
многих клиниках мира. Полученные результаты позволяют предполагать наличие
наследственной тромбофилии у любого пациента в том случае, если у него или его
кровного родственника имелись тромботические проявления в молодом возрасте.
В настоящее время обнаружены дефекты в генах следующих
факторов свертывания крови, приводящие к наследственным тромбофилиям:
. Антитромбин III (полное отсутствие или изменение
количества).
. Протеин С (полное отсутствие или изменение количества).
. Протеин S.
. Кофактор гепарина II.
. Фибриноген.
. Плазминоген.
. Активатор плазминогена (t-РА).
. Фактор Хакемана.
. Факторы тромбоцитов.
. Простациклин.
. АРС-резистентность фактора V.
. Гомоцистеинемия.
Причиной второго тромбофилического заболевания, возникающего
в результате наследования дефекта белка, был дефект протеина "Си".
Это явление было описано в 1981 г. Дж. Гриффином в США, а вскоре его наблюдали
в Голландии и Австрии. Причиной тромбозов явилась неспособность ограничивать
активность факторов V и VIII фибринообразования витамин К-зависимым белком,
протеином С. Этот дефект наблюдается в 6 - 8% случаев повторных тромбозов, у 3%
лиц, имевших первичный тромбоз глубоких вен, и у 0,2% здоровых лиц, т.е. в 10
раз чаще, чем дефект антитромбина III.
В 1984 г.П. Комп и С. Эсмон описали наследственную
предрасположенность к тромбозам в результате дефицита протеина S.
Оказалось, что данный белок, так же, как и протеин С,
участвует в ингибировании активных факторов V и VIII. Частота этого дефекта у
людей с тромбофилией колеблется по различным статистическим данным от 1 до 13%
и отмечается у 1 - 2% лиц с первичным тромбозом глубоких вен.
Эти работы заставили многих клиницистов думать о наличии
тромбофилий, однако более пристальное внимание к данной проблеме стало
оказываться лишь после 1993 г., когда шведский ученый Бьерн Дельбек сообщил о
семейной тромбофилии, возникшей в результате дефектного ответа плазмы больного
на добавление к ней активированного протеина С. Этот феномен, получивший
название "АРС-резистентность", был детально изучен многими авторами,
что привело к важному открытию. Оказалось, что фактор V свертывания крови, который
образуется в печени и мегакариоцитах, может иметь нарушение молекулярной
структуры, заключающееся в замене аргинина в положении 506 его полипептидной
цепи на глицин. Этот наследственный дефект оказался самым частым. У лиц с
частыми тромбозами он встречается в 52% случаев, у пациентов с первичным
тромбозом глубоких вен - в 20%, а среди здорового населения - в 3 - 7%. Весьма
интересным является тот факт, что данные дефекты чаще всего встречаются у
европейцев, а в азиатской популяции их частота уменьшается.
Согласно ранее приведенному списку, описаны случаи
тромбофилий, обусловленные дефектами фибриногена, плазминогена, ингибитора
тканевого активатора плазминогена, гепарин-кофактора II.
Гепарин-кофактор II был впервые описан в 1974 г. Бригиншоу и
Шанберг и выделен в 1981 г. Толлефсеном и др., а в 1981 г. - двумя группами
авторов - Траном, Марбелем и Дукеном, а также Силом и др. Брно и соавт. описали
случаи тромбофилий у лиц, имеющих врожденное снижение уровня гепарин-кофактора
II. Предполагают, что этот фактор обладает мощным антитромбиновым свойством и
может активироваться дерматаном-сульфатом на поверхности стенок сосудов, что
позволяет некоторым авторам рассматривать его в качестве "защитной системы
второго эшелона".
Протеин S осуществляет свое действие, опосредуя прикрепление
протеина С к мембранам тромбоцитов, что позволяет последнему взаимодействовать
с активированными факторами Va и VIIIA, а также с комплексом факторов Ха - Va.
Усиление фибринолитической активности при этом происходит за счет уменьшения
активности ингибитора РАI-1.
Одним из внешних признаков, позволяющих предполагать у
больных наличие дефицита протеина С, является появление кожных поверхностных
некрозов, особенно в тех местах, где есть избыток жировой ткани (липома). Это
особенно характерно для лиц, принимавших оральные антикоагулянты.
Причиной тромбозов как следствия наследственной тромбофилии
могут быть дефекты молекулы фибриногена. Любопытно, что дисфибриногенемии могут
быть причиной не только тромбозов, но и геморрагий. Описаны генетические
дефекты, проявляющиеся в нарушении структуры любой из трех цепей фибриногена
Аa, Вb и y-цепи. Связь между дефектами молекулы и фенотипом тромбозов пока не
ясна. Частота их невелика - 0,8% среди имевших тромботические явления.
У 19% лиц с ювенильными венозными тромбозами обнаруживают
гипергомоцистеинемию. В большинстве случаев этот фенотип наследуем.
Частота тромбозов при тяжелой гипергомоцистеинемии изучается,
хотя этот феномен встречается лишь у 1 на 300 000 жителей. Часты сообщения о
том, что тромботические ситуации сочетаются с дефектом плазминогена и
дисплазминогенемией. В то же время частота этой патологии составляет лишь 0,4%
в популяции, а у лиц с тромбозами - 3%. Все изложенное заставляет вести
детальные поиски причины тромбозов в каждом конкретном случае.
Повышенный риск венозного тромбоэмболизма, связанного с
наличием у больного дефектного аллеля соответствующего гена, доказан для
наследственного дефицита антитромбина III, протеина С, протеина S и фактора V,
что в сумме является причиной более 50% случаев наследственных тромбофилий.
Наследственные дефициты этих протеинов выявляют не только у лиц с тромбофилией,
так как они могут протекать и без клинических проявлений. Эти факты позволяют
предполагать, что дефицит лишь по одному протеину не всегда является
достаточным условием развития тромбофилии и для формирования соответствующего
фенотипа необходимо участие других факторов. Сочетание дефектов перечисленных
факторов является нередким, и обычно это сочетание характеризуется более
тяжелыми тромбофилиями, чем одиночные дефекты.
При изучении тромботического и геморрагического анамнеза у 77
лиц с наследственным дефицитом фактора XII Б. Дэммле и соавт. установили, что
тромботические осложнения имелись лишь в 3 случаях, в то время как склонность к
кровоточивости отмечена у 1 больной, у которой уровень фактора XII был в
пределах 50% от нормы (XII: С - 52%, XII-Ar - 53%). Наблюдая за эффективностью
профилактического назначения оральных антикоагулянтов у больных с дефицитом
антитромбина III, протеина С, плазминогена и дисфибриногенемией, Жд. Финацци и
соавт. (1989) не отметили тромбозов глубоких вен при уровне протромботического
времени ИНР - 2,5 - 4,0; наблюдения за пациентами с дефицитом фактора V выявили
случай рецидивирующего тромбоза вен нижних конечностей с ТЭЛА артерии у
больного, имевшего снижение коагуляционного уровня фактора V и антигена фактора
V соответственно до 12 и 10%.
Нефротический синдром также сопровождается тромботическими
осложнениями в связи с тем, что гипоальбуминемия не позволяет формироваться
фибриновому сгустку, к которому имеется тропность у плазминогена, и создавать
условия контактирования плазминогена с его тканевым активатором.
Профилактика и лечение наследственных тромбофилий не являются
чем-то особенным и могут успешно осуществляться имеющимися на вооружении
клиницистов гепарином и оральными антикоагулянтами. В связи с этим основной
проблемой этой части современной медицины являются выявление маркеров
тромбофилий и отработка режимов противотромботической терапии (дозировки препаратов
и длительности их назначения).
Можно предполагать, что предрасположенность к тромбозам
детерминирована генетически во всех случаях их возникновения - и при поражении
сосудов (в том числе и атеросклерозом), и при недостаточности кровообращения. Однако
сегодня с достоверностью можно говорить о наличии генетических дефектов -
мутаций в генах системы свертывания крови - лишь при этом типе тромбофилий.
Мутационные изменения в генах могут происходить на протяжении
всей жизни человека. В том случае, если мутации возникают в геноме клеток
зародышевой линии, все соматические клетки организма-потомка, развивающегося из
мутантной зиготы, образовавшегося от слияния мутантных яйцеклетки и
сперматозоида, будут содержать указанную мутацию. Чем позже в онтогенезе
(индивидуальном развитии) человека возникает соматическая мутация, тем меньше
размер клона мутантных клеток, заключенного во взрослом организме. Если же
мутация доминантна, т.е. патологический мутантный признак, определяемый
мутантным геном, проявляется даже при наличии в соматических клетках копии
нормального гена, полученного от другого родителя, развивается генетическое
наследственное заболевание.
В случаях, когда мутация, определяющая мутантный фенотип,
рецессивна (ее действие проявляется лишь в гомозиготном состоянии, при котором
один и тот же мутантный ген получен от обоих родителей), можно говорить о
предрасположенности организма к соответствующему заболеванию и носительстве
мутантного гена. Организм, у которого действие рецессивной мутации маскируется
функционированием другого полноценного аллеля, внешне (фенотипически) выглядит
совершенно нормальным. Однако у него гораздо больший риск дать больное
потомство в браке с носителем такого же мутантного гена, что является одной из
причин запрета на близкородственные браки. С другой стороны, у носителей
определенных мутантных генов в гетерозиготном состоянии может произойти
соматическая мутация в соответствующем аллельном гене соматических клеток, что
также станет причиной развития приобретенного генетического заболевания. Как
известно, гены человека заключены в молекулах ДНК его хромосом, а изменения
структуры ДНК, называемые мутациями, нарушают функционирование генов. В связи с
этим выявление мутаций в конкретных генах человека является наиболее точным и прямым
методом диагностики генетических заболеваний.
В основе всех методов ДНК-диагностики лежат три
фундаментальных принципа молекулярной биологии. Это прежде всего существование
комплементарных взаимодействий между двухцепочечными молекулами нуклеиновых кислот
(первый принцип), которые после денатурации с последующей ренатурацией (второй
принцип) позволяют им безошибочно находить друг друга и восстанавливать
первоначальную структуру. Если же в процессе ренатурации нуклеиновых кислот
добавить в пробы короткие молекулы одноцепочечной ДНК (зонды или праймеры),
ковалентно соединенные с каким-либо меченым соединением (третий принцип), то
они благодаря комплементарным взаимодействиям соединяются с тем участком
нуклеиновой кислоты, который содержит последовательность нуклеотидов, строго
соответствующую последовательности нуклеотидов зонда (или праймера). Наличие
связавшейся с нуклеиновой кислотой метки с высокой точностью свидетельствует о
присутствии в анализируемом образце искомых последовательностей нуклеотидов.
Разработанные недавно методы молекулярной биологии и особенно
метод полимеразной цепной реакции (ПЦР) значительно облегчили проведение
диагностики наследственных заболеваний. При этом мутации выявляются
непосредственно в ДНК клинических образцов, чаще всего крови. Суть этой реакции
заключается в том, что короткие фрагменты исследуемого гена (праймеры),
добавленные к анализируемой ДНК, специфически связываются с поврежденным
участком исследуемого гена и делают возможным многократное его воспроизведение
с помощью специального фермента - ДНК-полимеразы. Образующийся при этом продукт
ПЦР может быть легко обнаружен с помощью электрофореза. Его отсутствие отрицает
искомую патологию.
Полиморфизм
генов тромбофилии
Гены белков противосвертывающей системы крови

Рис. 1. Роль белка PAI-1 в процессе фибринолиза
Для нашей работы представляет интерес инсерционно-делеционный
4G/5G полиморфизм (rs1799889 (-)): выпадение одного из пяти
гуаниновых нуклеотидов в (-675) позиции промоторной области. Аллель 5G имеет меньшую
транскрипционную активность, наличие мутантной аллели 4G приводит к повышению
активности PAI-1 и склонности к тромбозам [55]. По результатам нескольких
исследований показано, что пациенты с аллелью 4G имеют большие шансы
тромботических осложнений, чем пациенты с генотипом 5G/5G [24] [26] [57]. Частоты
генотипов для европейской популяции: NN - 0.261; NM - 0.391; MM - 0.348. Частоты аллелей
для европейской популяции: N - 0.457; 0.618; M - 0.543; 0.382 [66]
Последние исследования показали, что данный полиморфизм
ассоциирован с тяжелым течением нефрита у больных СКВ. У пациентов с генотипом MM уровень протеинурии был
выше, чем у больных без мутации (NN) и носителей (NM). По результатам биопсии
у пациентов с мутантным генотипом отмечались более тяжелые морфологические
поражения почек. [64] Показано, что мутация в гене PAI-1 является
дополнительным фактором риска развития ИМ и ИБС у больных СКВ [34]. Аллель 4G также повышает риск
развития тромбозов глубоких вен голеней [15] [24], атеросклероза [5], ИМ [16] и
асептического некроза головки бедренной кости [30].
Гены белков свертывающей системы крови
Ген
фактора XIII свертывания крови (G485T)
Фибринстабилизирующий фактор, - профермент, активируемый
последним в гемокоагуляционном каскаде. Принадлежит к семейству
трансглутаминаз, которые катализируют образование специфических поперечных
ковалентных связей между полимерными нитями фибрина, между фибрином и
некоторыми плазменными белками, а также белками внеклеточного матрикса [59]
(рис.2)

Рис. 2 Участие FXIII в процессе полимеризации фибрина
Из полиморфизмов, не приводящих к дефициту цепи А белка,
стоит отметить однонуклеотидную замену G на Т во втором экзоне
гена цепи А, которая приводит к замене валина на лейцин в 34 позиции
аминокислотной последовательности активационного пептида белка (V34L), что приводит к более
быстрой активации мутировавшей изоформы фактора XIII, а это, в свою очередь,
приводит к образованию более прочного и стабильного тромба [31] [42].
Частота генотипов и аллелей в европейской популяции - NN - 45,2%; NM - 44,9%; MM - 9,9%; N - 67,2%; M - 32,8% [66]
Обнаружено, что у пациентов без ИМ аллель 34L встречается значительно
чаще, чем у больных [63]. Также выяснено, что риск развития венозных тромбозов
также снижается у пациентов с аллелем 34L [24]. Это указывает на
то, что данный полиморфизм является протективным по отношению к развитию ИМ и
возможно тромбозов. Механизмы связи полиморфизма гена и его протективного
действия не ясны. Роль данного полиморфизма в развитии тромботических
осложнений и атеросклероза остается дискутабельной [42].
Гены тромбоцитарных рецепторов
Ген
гликопротеина Ia (GPIa) (C807T)
Гликопротеин Iа или α2β1 - интегрин, - основной
рецептор коллагена на тромбоцитах. Состоит из гликопротеинов Ia (α2-субъединица) и IIа (β1-субъединица) Обеспечивает
адгезию тромбоцитов как к фибриллярным так и к нефибриллярным типам коллагена.
Этот гликопротеин экспрессируется в относительно небольших количествах (1000 -
3000 копий на тромбоцит), но по разным данным плотность рецепторов у разных
людей может отличаться в 10-40 раз и пропорциональна степени адгезии
тромбоцитов к коллагену [11] [65]. Уровень экспрессии данного белка связан с
полиморфизмами гена α2 субъединицы.
Однонуклеотидная замена цитозина на тимин в позиции 807 не приводит к изменению
аминокислотной последовательности белка, но влияет на уровень экспрессии (т. н.
silent polymorphism). Аллель M ассоциируется с
повышенной плотностью рецепторов на тромбоцитах и повышенной адгезией
тромбоцитов к коллагену 1 типа [14]. Частота генотипов и аллелей в европейской
популяции - NN - 47%; NM - 49%; MM - 4%; N - 70%; M - 30% [66]
Повышенная плотность рецепторов на
мембране тромбоцита увеличивает риск развития тромботических осложнений.
По некоторым данным, риск развития тромбозов у молодых пациентов при наличии
аллели Т возрастает в 2-3 раза, особенно в случаях с отягощенным развитием
сахарного диабета [47]. Также показано, что у гомозигот по аллели Т в 7 раз
увеличивается риск повторных приступов острого коронарного синдрома [5].
Изучаемый полиморфизм связан с повышенным числа проявлений артериальных и
венозных тромбозов у больных с АФС [65].
Ген
гликопротеина IIIa (GPIIIa) (T196C)
Гликопротеин IIIa (β3-интегрин) является
субъединицей рецептора фибриногена - гликопротеина IIb/IIIa на поверхности мембраны
тромбоцитов. Данный гликопротеин - наиболее распространённый трансмембранный
рецептор тромбоцитов (60 000 - 100 000 копий на клетку и 1% -2% общего
количества белков тромбоцита), его активация является ключевым, процессом
запускающим агрегацию тромбоцитов [20]. Предполагается также, что β3-интегрины играют важную роль не только в тромбообразовании, но и в
процессах миграции и пролиферации клеток [45]. В гене идентифицировано около 10
однонуклеотидных полиморфизмов, в т. ч. PLA1 и PLA2. Аллель PLA2 представляет собой
результат замены тимидинового нуклеотида на цитозиновый в позиции 196 третьего
экзона. В результате происходит замена лейцина (норма при PLA1) на пролин (изменение
при PLA2) в 33 аминокислотном остатке, что производит к изменению
комформации белка и пространственной ориентации лиганд-связывающего участка
[11].
Частота генотипов и аллелей в европейской популяции - NN - 72%; NM - 26%; MM - 1,6%; N - 96%; M - 4% [66]
Носительство генотипа MM, по данным литературы,
увеличивает риск развития ИБС и ИМ в 2-4 раза [23] [14] [2], а в сочетании с
гиперхолестеринемией в 6 раз [8].
Гены белков, вовлеченных в патогенез
эндотелиальной дисфункции
Ген
P22 phox субъединицы NADPH-оксидазы (C242T)
Основным источником радикалов кислорода в сосудистой стенке,
как и в фагоцитах, является фермент NADPH-оксидаза, которая участвует в процессе
катализации кислорода. При активации NADPH-оксидазы нарушается выработка NO, вазодилатирующая
способность NO снижается, в эндотелии сосудов поддерживается оксидантный стресс,
происходит структурная перестройка, как эндотелия, так и гладкомышечных клеток
сосудов (рис. 3) [7] [62]. Кроме того, активация NADPH-оксидазы способствует
стимуляции симпатической активности и повышению артериального тонуса [29].
Таким образом, активация NADPH-оксидазы и снижение продукции оксида азота
способствует становлению АГ, и развитию осложнений, таких как хроническая
сердечная недостаточность, атеросклероз, нефропатия [38].

Рис. 3 Участие NAD (P) H оксидазы в патогенезе эндотелиальной дисфункции.
Ген CYBA, кодирующий p22 субъединицу,
локализован на хромосоме 16q24 и существует в нескольких аллельных вариантах. Одним из
таких полиморфизмов является однонуклеотидная замена цитозина на тимин (С242Т),
приводящая к замене гистидина на тирозин в 72 позиции аминокислотной
последовательности p22-phox, которая определяет активность NADPH-оксидазы [58]. Выло
выявлено, что в клетках, экспрессирующих 242Т p22-phox, активность продукции
кислородных радикалов NADPH-оксидазой значительно выше, чем в клетках не
экспрессирующих 242Т p22-phox субъединицу [62].
Частота генотипов и аллелей в европейской популяции - NN - 38%; NM - 46%; MM - 15%; N - 61%; M - 39% [66]
У носителей генотипа MM шанс возникновения ИБС и
атеротромботических осложнений повышается в 2-3 раза [39] [44].
Ген
эндотелиальной NO синтазы (G894T)
Оксид азота синтезируется из L аргинина посредством
ферментов семейства NO-синтаз. В настоящее время большой интерес представляет изучение
генов, ответственных за уровень NO и их влияние на развитие нарушений функций
эндотелия и опосредованных ими тромботических осложнений. Одним из таких генов
является ген эндотелиальной NO-синтазы (eNOS)
Ген eNOS локализован в хромосоме
7q35-36 и кодирует белок,
состоящий из 1203 аминокислот. Наиболее мощным регулятором экспрессии eNOS является напряжение
сдвига, связанное с воздействием потока крови на поверхность эндотелиальных
клеток [38].
Лучше всего изученным и прогностически значимым является
структурный полиморфизм экзона 7 - замена гуанина тимином в 894-й позиции гена,
что приводит к замене глутамина аспарагином в 298-й позиции фермента. Эта
мутация, вероятно, вызывает конформационные изменения в кодируемом белке [33].
Частота генотипов и аллелей в европейской популяции - NN - 58%; NM - 35%; MM - 7%; N - 76%; M - 24% [66]
При исследовании нативных сосудов показано
снижение каталитической активности у носителей eNOS 894T [7]. Показано, что эта
мутация является фактором риска развития венозных тромбозов. Особый интерес к
данному полиморфизму вызывает тот факт, что аКЛ дополнительно снижают
активность eNOS [49].
Ген
метилентетрагидрофолатредуктазы (MTHFR) (С677Т)
За последние годы проведено множество исследований,
указывающих на независимое участие гипергомоцистеинемии в развитии
тромботических осложнений. Механизмы влияния гомоцистеина на процесс развития
тромбозов в значительной степени связаны с эндотелиальной дисфункцией.
Показано, что гипергомоцистеинемия приводит к повреждению
эндотелия различными способами. В эндотелии образуются повреждения, обнажающие
субэндотелиальный матрикс и гладкомышечные клетки, изменяется направление их
пролиферации, что способствует активации тромбоцитов и лейкоцитов и увеличению
тромбоцитарной активности [22]. Известно также, что гомоцистеин меняет свойства
эндотелия, активируя фактор XII и фактор V, а также экспрессию
тканевого фактора, подавляя активность тромбомодулина и гепаринового сульфата.
Все это способствует формированию тромбина [41]. Наряду с описанными эффектами
гомоцистеин может компенсировать выделение эндотелиального оксида азота,
который соединяется с гомоцистеином в присутствии кислорода. Гомоцистеин может
также уменьшать синтез оксида азота благодаря эффекту инициации перекисного окисления
липидов, которое уменьшает экспрессию синтазы оксида азота и тем самым
непосредственно снижают синтез NO [7]. В 1995 г. Fross и соавт. описали частый
полиморфизм, заключающийся в замене цитозина на тимин в 677-м положении гена
(С677T),
кодирующего MTHFR, фермент участвующий в метаболизме гомоцистеина [11] (рис.4).
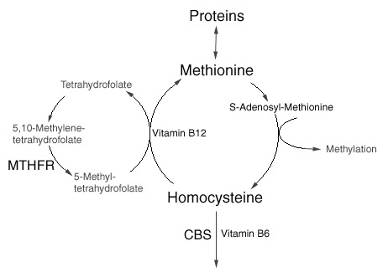
Рис. 4 Участие MTHFR в метаболизме гомоцистеина
В результате замены происходит замена Glu на Ala в первичной структуре
белка, что в свою очередь приводит к образованию термолабильного и менее
активного варианта фермента. Это ведет к уменьшению образования
5-метилтетрогидрофолата и соответственно метионина из гомоцистеина и накоплению
последнего [41]. Частота генотипов и аллелей в европейской популяции - NN - 56%; NM - 36%; MM - 9,9%; N - 74%; M - 26% [66]
Показано, что наличие мутации в гене MTHFR ассоциировано с
развитием тромбозов и как следствие более тяжелого течения ИБС [5].
Носительство аллели Т не вызывает развитие тромботической и сердечно-сосудистой
патологии само по себе, а только в сочетании с традиционными факторами риска,
такими как АГ и ожирение [11]. У пациентов с венозными тромбозами мутация в
данном гене ведет к повышению риска развития ТЭЛА [1]
Кардиоренальный
синдром
Существуют разные определения кардиоренального синдрома:
сочетание сердечной и почечной недостаточности [4,5], патофизиологическое
состояние, при котором сочетание дисфункции сердца и почек усугубляет
недостаточность каждого органа, повышая летальность при той и другой патологии
[6,7], сердечная недостаточность, осложненная почечной дисфункцией [8]. Н.А.
Мухин и соавт. [9] патологию сердца и почек при ишемической болезни почек
(атеросклеротической реноваскулярной гипертонии) трактуют как кардиоренальный
синдром.
С. Ronco и соавт. [1] предложили делить кардиоренальный
синдром на типы. Тип 1 КРС - тяжелое нарушение функции сердца (кардиогенный шок
или декомпенсация хронической сердечной недостаточности) ведет к острому
почечному повреждению. Тип 2 КРС - хроническая сердечная недостаточность
приводит к ухудшению функции почек (хронической болезни почек). Тип 3 КРС -
быстрое ухудшение функции почек (ишемия почек, гломерулонефрит) ведет к острой
дисфункции сердца (аритмия, острая сердечная недостаточность). Тип 4 КРС -
хроническая болезнь почек приводит к гипертрофии миокарда, снижению функции
сердца и увеличению риска сердечно-сосудистых осложнений. (Шутов А.М., Серов В.
А, 2009)
КРС 5 типа (вторичный КРС) - состояние, при котором системная
патология приводит к сочетанной сердечной и почечной дисфункции. Спектр
состояний, которые одновременно приводят острому/хроническому патологическому
взаимодействию сердце-почки, чрезвычайно разнообразен: системные и инфекционные
заболевания, опухоли, осложнения лекарственной терапии, амилоидоз, сахарный
диабет и т.д. Точных данных об эпидемиологии данного варианта КРС нет.
Механизмы его развития сложны и требуют уточнения. В связи с этим лечение на
сегодняшний день заключается в воздействии на основную причину заболевания [6].
Изучение причин и механизмов формирования данного типа КРС, раннее выявление
биомаркеров повреждения и факторов риска поможет определить оптимальные методы
коррекции с целью улучшения выживаемости и повышения качества жизни пациентов.

Основные
патофизиологические механизмы формирования кардиоренального синдрома
В настоящее время концепция кардиоренального синдрома
основана на существовании взаимно влияющих патогенетических факторов, как
оказывающих неблагоприятное влияние в отношении сократительной способности
миокарда, так и определяющих прогредиентное снижение функциональной способности
и выживаемости почки. Причем именно доказанная синергичность указанных
взаимоотношений, опосредованная вовлечением в патологический процесс разнообразных
генетических, гемодинамических, метаболических, структурно-функциональных
факторов, нейрогуморальной и провоспалительной активацией, нарушениями
липидного и минерального обмена, привела к появлению термина
"кардиоренальный синдром" [9, 53, 57]. В качестве прекурсоров и
предрасполагающих к появлению последнего факторов обычно рассматривают
артериальную гипертензию (АГ), анемию, гипер - и дислипидемию, ишемическую
болезнь сердца, сахарный диабет, метаболический синдром/абдоминальное ожирение,
ХБП, реноваскулярные заболевания [63, 64, 69].
При этом в качестве основных механизмов, способствующих
формированию кардиоренального синдрома, рассматривают повышение центрального
венозного давления, снижение перфузии почки, внутрипочечную гипертензию,
недостаточность эндогенных механизмов, обеспечивающих эффективный салурез и
натрийурез (система натрийуретических пептидов), эндотелиальную дисфункцию,
системную провоспалительную и нейрогуморальную активацию, прооксидантный стресс
и некоторые другие факторы (рис. 2).

Фактически критическое снижение тканевой перфузии
органа-мишени с последующим возникновением ишемического (реперфузионного)
воспалительного повреждения является морфологической основой кардиоренального
синдрома.
На сегодняшний день большинство исследователей разделяют
следующую точку зрения на патогенез острого повреждения почек при ишемических
атаках в сердце. Основной особенностью ответной реакции почек на падение
перфузионного давления является ауторегуляция - поддержание нормальных
кровотока и скорости клубочковой фильтрации даже при среднем артериальном
давлении равном 80 мм рт. ст. Ауторегуляторная реакция при гипоперфузии
заключается в первую очередь в снижении сосудистого сопротивления афферентных
артериол, медиаторами которого выступают простагландины. Помимо этого, давление
в клубочковых капиллярах, а именно оно является движущей силой процесса
фильтрации, поддерживается увеличением тонуса эфферентных артериол. За данную
фазу ауторегуляции ответственны вазоконстрикторные субстанции, в частности
ангиотензин II [5]. Приведенная схема касается случаев, когда процесс
ауторегуляции не нарушен. Однако во многих случаях защитные механизмы не
срабатывают, и гипоперфузия почек запускает каскад взаимосвязанных патологических
реакций, что в конечном итоге приводит к острой почечной недостаточности, даже
при нормальном уровне артериального давления [4].
Подобное развитие событий можно наблюдать у пациентов с
распространенным атеросклерозом, артериальной гипертензией, когда гиалиноз и
миоинтимальная гиперплазия вызывают структурное сужение артериол [20].
Некоторые специалисты связывают невозможность адекватного снижения сосудистого
сопротивления с истощением эндогенных сосудорасширяющих веществ.
Вазоконстрикторы, секретируемые в кровь при состояниях, сопровождающихся
гипоперфузией почек, действуют практически без всякого ограничения, которое в
норме обеспечивали бы эндогенные вазодилататоры, например, простагландины.
После завершения этапа первичного повреждающего действия гипоперфузии
запускается активация различных гормональных систем, действие которых,
направленное на компенсацию сниженного кровотока, еще больше усугубляет
почечную недостаточность. Наиболее активным действующим агентом в данной
ситуации, большинство исследователей считают ангиотензин II [23], реализующий
свой основной эффект через повышение сосудистого сопротивления выносящих
артериол. С увеличением тяжести и продолжительности ишемии увеличивается
вероятность перехода процесса в следующую стадию - структурного повреждения
канальцев, что еще больше усугубляет ухудшение функции почек. Поврежденные
клетки эпителия, а также остатки апикальной мембраны скапливаются в просвете
канальцев и формируют цилиндры, которые закупоривают просвет, вызывая феномен
"обратного тока" клубочкового фильтрата через поврежденный эпителий в
кровеносное русло. Нарушенная реабсорбция калия ведет к увеличению его
концентрации внутри канальца, что запускает полимеризацию белка
Тамма-Хорсфолла, который в норме секретируется в петле Генле, превращая его в
гель и также способствуя обструкции просвета канальца [8]. Усугубляющаяся
ишемия и истощение запасов кислорода приводят к полному израсходованию
внутриклеточного АТФ, что способствует дальнейшим критическим изменениям
метаболизма в клетках эпителия. Нарушения в энергетическом обмене выражаются в
постепенной дислокации белков цитоскелета, разрушении межклеточных соединений,
в результате чего остатки клеток эпителия в еще большем количестве устремляются
в просвет канальца, загромождая его просвет. Огромное значение при ишемии имеет
баланс между эндогенными вазодилатирующими веществами, к которым можно отнести
оксид азота, с одной стороны и циркулирующими радикалами кислорода, с другой.
Большим количеством работ подтверждено, что различные формы радикалов кислорода
вызывают сосудосуживающий эффект, оказывают угнетающее действии на натрийурез и
способствуют задержке жидкости [7]. При ишемических явлениях в любых тканях
организма соотношение кислородных радикалов и эндогенных вазодилататоров резко смещается
в сторону первых, вызывая так называемый оксидативный стресс [38]. Почечная
ткань не является исключением из данного правила. Помимо оказываемого
сосудосуживающего эффекта, активные формы кислорода активируют протеазы и
фосфолипазы, вносящие огромный вклад в разрушение клеточных структур, молекул
ДНК, белков, липидов и углеводов, которые в свою очередь, являются активными
медиаторами воспаления, вызывая миграцию к месту повреждения лейкоцитов и
макрофагов [8]. Важную роль в запуске воспалительной реакции играет ангиотензин
II, реализующий свой провоспалительный эффект через сигнальную молекулу
нуклеарный фактор Каппа B, которая запускает продукцию хемокинов и молекул
адгезии. После завершения всех вышеописанных процессов наступает фаза
репарации, которая чаще всего завершается очаговым гломерулосклерозом и
компенсаторной гиперфункцией оставшейся почечной ткани [7].
Наиболее точным показателем, отражающим функциональное
состояние почек, является скорость клубочковой фильтрации (СКФ). Для оценки
скорости клубочковой фильтрации были разработаны два варианта формулы MDRD
(Modification Diet in Renal Disease): полная и сокращенная. Для расчета СКФ по
полной (оригинальной) формуле требуется ряд биохимических показателей сыворотки
крови: альбумин, азот мочевины и креатинин. Для использования сокращенной
формулы MDRD необходимы только демографические данные (пол, возраст, раса) и
уровень креатинина сыворотки. Формула MDRD (мл/мин/1,73 м2) СКФ = 186 x
(креатинин сыворотки, мг/дл) - 1,154 × (возраст, годы) - 0, 203
для женщин результат умножают на 0,742, для лиц негроидной расы результат
умножают на 1,210.
Результаты, получаемые при применении обеих формул,
сопоставимы. Несмотря на некоторую неточность данной формулы при оценке функции
почек у пациентов с сердечно-сосудистыми заболеваниями, СКФ, рассчитанная
данным методом, является независимым предиктором летального исхода у больных с
дисфункцией левого желудочка [14]. При применении формулы MDRD значения СКФ ≤
60 мл/мин/1,73 м2 считаются патологическими (уровень доказанности I, В) [26].
Для скрининга на наличие почечного повреждения рекомендуется использовать
отношение Альбумин/Креатинин в моче, которое принято считать патологическим при
превышении 30 мг/г (уровень доказанности IIа, В) [9,27].
Материалы и
методы
В исследование отбирались больные обоего пола с установленным
диагнозом СКВ, европеоидной расы, наблюдающиеся в клинике нефрологии,
внутренних и профессиональных болезней им.Е.М. Тареева. Длительность наблюдения
составляла для 73 больных 5 и более лет, для 18 больных от 1 года до 5 лет и
для 9 больных 1 год и менее. Диагноз СКВ ставился согласно критериям
Американской коллегии ревматологов [4]. Основанием для диагноза АФС служили,
разработанные и дополненные группой исследователей во главе с D. Alarcon-Segovia
в 1999 году, клинико-лабораторные критерии [3] [60]. Диагноз считался
подтвержденным при наличии одного клинического и одного лабораторного критерия.
В работе АФС был выявлен у 49 (49%) пациентов.
Общее количество пациентов, задействованных в исследовании,
составило 100 человек в возрасте от 16 до 73 лет (средний возраст - 37лет (СО:
14 лет), из них 80 женщин (80%) и 20 мужчин (20%).
Всем больным проводилось стандартное для больных СКВ
обследование. Применялись комплексные клинико-лабораторные и инструментальные
методы исследования.
Клинические данные анализировались на основании историй
болезней пациентов.
Пациенты были разделены по значению СКФ на 2 группы: 1-я
группа - с СКФ более 60 мл/мин/1,73 м2; 2-я - с СКФ менее 60 мл/мин/1,73 м2.
Значения СКФ, оцененной по формуле MDRD, у обследованных
пациентов распределены следующим образом: нормальное значение СКФ
констатировано у 39 (39 %) обследованных; умеренное снижение СКФ наблюдалось
также у 39 (39 %); ХБП 3-й стадии - у 18 (18 %); ХБП 4-й стадии - у 3 (0,29 %);
ХБП 5-й стадии - у 2 (2%) пациентов. Таким образом, ориентируясь на показатель
СКФ, рассчитанной по формуле MDRD, ХБП была диагностирована у 22 (22 %)
больных.
В работе больные сравнивались по наличию и отсутствию
волчаночного нефрита. Волчаночный нефрит был выявлен у 74 (74%) пациентов.
Проводилась дифференцировка больных в зависимости от
клинических форм ВН. Клинические формы ВН определяли по классификации И.Е.
Тареевой (1976 г).
Всем больным определялись гены тромбофилии PAI-1, Nos, P22,
MTHFR, Gp-Ia, Gp-IIIa, FXIII.
Анализ полиморфизмов генов проводился с помощью
аллельспецифичной полимеразной цепной реакции и электрофореза в агарозном геле.
Определение полиморфизмов генов проводилось на базе
лаборатории генных и клеточных технологий кафедры биохимии Факультета
фундаментальной медицины Московского государственного университета им. М.В.
Ломоносова.
Методы
статистической обработки
Обработка данных проводилась с помощью пакета программ Statistica 10. При оценке и анализе
полученных данных применялись стандартные методы описательной статистики. Для
количественных признаков, имеющих нормальное распределение значений,
вычислялось значение средней величины и ее стандартного отклонения. Для
качественных признаков, значимых в сравнении групп, вычислялась относительная
частота и стандартная ошибка доли.
Определение статистической значимости различий между группами
пациентов проводили с использованием критерия χ2 (по методу Пирсона и
методу максимального правдоподобия), Статистически значимыми считались различия
при p < 0,05. При значимых различиях, дополнительно проводилось вычисление
отношения шансов.
Результаты
исследования

Рис. 3. Распределение больных по стадиям ХБП

больные СКВ популяция
Рис. 4. Спектр ССО в разных возрастных группах у больных с
СКВ и в популяции

Рис. 5. Спектр ССЗ у больных СКВ

Рис. 6. Спектр ССО у больных с кардиоренальным синдромом в
зависимости от пола

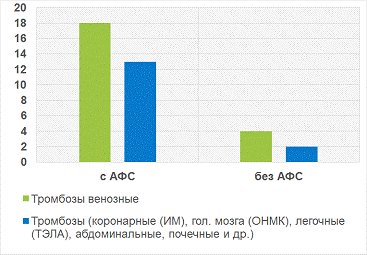
Рис. 8. Варианты тромботических осложнений у больных с КРС

Рис. 9. Частота волчаночного нефрита у больных СКВ

Рис. 10. Частота волчаночного нефрита у больных СКВ
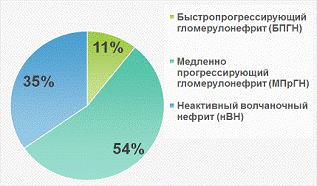
Рис. 11. Частота встречаемости форм волчаночного нефрита у
больных СКВ

Рис. 12. Частота нарушения функции почек в зависимости от
типа ВН

Рис. 13. Варианты ССО в зависимости от формы ВН у больных с
нарушенной функцией почек
Обсуждение
результатов исследования
В нашем исследовании количество больных, имеющих высокий и
очень высокий риск сердечно-сосудистых осложнений, в сумме составило 22
человека (22%). У данной группы пациентов нарушения функции почек варьировали
от умеренного снижения СКФ до терминальной почечной недостаточности.
У пациентов с СКВ, при которой по разным данным в 50-70%
развивается поражение почек, СС патология превалирует в молодом возрасте.
Поражение сердца по нашим данным выявлено у трети больных моложе 40 лет. Более
чем у половины пациентов в молодом и среднем возрасте от 40 до 59 лет
встречаются различные варианты поражения ССС. Что значимо отличается от
популяционных показателей в тех же возрастных группах. Поэтому важным
представляется изучение структуры СС заболеваемости и типов поражения ССС в
рамках СКВ, сочетанных с поражением почек (КРС).
У больных с кардиальной патологией были выявлены в основном
поражения по типу АГ. При сравнении групп больных с выраженным нарушением
функции почек и больных с ХБП 1-2 не выявлено статистически значимых различий в
частоте проявлений различных вариантов поражения ССС.
У женщин чаще наблюдалась кардиопатия (что можно трактовать в
рамках СКВ), в то же время сосудистая патология наблюдалась чаще у мужчин
(мужской пол является независимым фактором риска развития сосудистых событий).
При сочетании АФС и КРС - количество пациентов с тяжелыми ССО
(тромбозы, ОКС, АГ 3 ст 3степ, стенокардия 3-4ФК), было выше (5), чем при
солидном течении КРС (тяжелые осложнения наблюдались лишь у одного пациента).
Что позволяет считать АФС фактором риска тяжелого течения поражения сердца у
больных СКВ, в сочетании с патологией почек.
В рамках АФС частота артериальных и венозных тромбозов
значимо выше, чем у пациентов без данного синдрома. Тромботические осложнения
очень часто приводят к серьезным поражениям ССС, ухудшают прогноз течения
заболевания в целом и требуют более агрессивной тактики лечения.
У 74% пациентов по нашим наблюдениям развивались те или иные
формы волчаночного нефрита.
Сочетание поражения ССС и почек наблюдалось у 38% больных.
Доказанный КРС был выявлен лишь у 8 пациентов.
Наиболее прогностически опасной формой волчаночного нефрита
является БПВН, у таких пациентов нарушение функции почек встречается в 38%
случаев. Наиболее распространенный вариант поражения почек при СКВ МПВН -
нарушение почечной функции отмечается у 27% пациентов. Наиболее "легкая"
форма поражения почек при СКВ - неактивный ВН, при этом у незначительного числа
пациентов наблюдается ухудшение функции почек, чаще всего связанное с дебютом
заболевания с активного почечного процесса, но в дальнейшем в результате
лечения у таких больных удалось добиться контроля над показателями креатинина
(в нашей работе не более 2,2 мг/дл) и протеинурией (чаще всего следовые
значения).
Среди всех типов нефрита кардиопатия наиболее часто
встречается у пациентов с БПВН (18%) и нВН (20%) Аналогичное распределение наблюдается
в отношении тромботических осложнений, при БПВН (36%), при НВН (40%). (в случае
с БПВН такое сочетание обусловлено возможно общей остротой процесса, при нВН
вероятно по причине хронизации поражения почек). Артериальная гипертензия,
обусловленная поражением почек, чаще всего ассоциирована с МП формой ВН (63%).
У всех больных с доказанным кардиоренальным синдромом (8)
были выявлены различные варианты полиморфизма генов тромбофилии.
• У двух пациентов
присутствовали мутации в гене MTHFR
Клинически у этих пациентов в первом случае - наблюдалась
неактивная форма ВН в сочетании с перикардитом, во втором - медленно
прогрессирующая форма ВН и панкардит.
• По одному пациенту имели
мутации в генах eNos и PAI-1
(полиморфизм eNOS был ассоциирован с быстропрогрессирующим ВН и
эндокардитом;
полиморфизм PAI-1 - с медленно прогрессирующим ВН и
миокардитом).
• Также стоит отметить, что
у одного пациента выявлены полиморфизмы одновременно в двух генах (FXIII, p22). При этом клинически
наблюдался медленно прогрессирующий ВН в сочетании с перикардитом.
Выводы
• У 38% пациентов с СКВ
наблюдается сочетание поражения ССС и почек. У 8% удалось достоверно установить
диагноз КРС (что связанно со сложностью точной интерпретации
причинно-следственной связи между поражением почек и ССС).
• Среди всех типов нефрита
кардиопатия наиболее часто встречается у пациентов с БПВН (18%) и нВН (20%).
Аналогичное распределение наблюдается в отношении тромботических осложнений
(при БПВН (36%), при НВН (40%). Артериальная гипертензия, обусловленная
поражением почек, чаще всего ассоциирована с МП формой ВН (63%).
• АФС является фактором
риска тяжелого течения ССЗ у больных СКВ в сочетании с почечным поражением.
• Наличие полиморфизма
генов тромбофилии MTHFR, FXIII, p22, eNos и PAI-1 ухудшает прогноз в отношении течения КРС у больных СКВ.
Список
литературы
1. Агеев
Ф.Т. Роль эндотелиальной дисфункции в развитии и прогрессировании
сердечно-сосудистых заболеваний. Сердечная недостаточность 2003; 4: 1: 22.
2. Алекперов
Р.Т., Тимченко А.В., Самсонов М.Ю. и др. Уровень растворимого рецептора 1 типа
фактора некроза опухоли у больных системной склеродермией. Тер арх 2004; 5:
11-15.
. Брундтланд
Г.Х. Открытие заседаний Научной группы ВОЗ по ущербу при мышечно-скелетных
заболеваниях. Научно-практ ревматол 2001; 1: 5-7.
. Вдовченко
Л.В., Марасаев В.В. Анализ причин смерти у больных ревматоидным артритом.
Научн-практ ревматол 2001; 3: 22.
. Демина
А.Б., Раденска-Лоповок С.Г., Фоломеева О.М., Эрдес Ш. Анализ структуры
летальных исходов и причин смерти при ревматических заболеваниях в Москве.
Научно-практ ревматол 2004; 2: 25-31.
. Каратеев
Д.Е. Основные тенденции и вариабельность эволюции ревматоидного артрита:
результаты многолетнего наблюдения. Научно-практ ревматол 2004; 1: 8-13.
. Комаров
В.Т., Девина О.В., Воеводина Т.С. Характеристика причин смертности при
ревматоидном артрите. Научно-практ ревматол 2003; 2: 46.
. Крикунов
В.П., Головизнин М.В. Прогрессирование ревматоидного артрита и клиническая
оценка вариантов его течения. Рос ревматол 1999; 5: 8-14.
. Моисеев
В.С., Кобалава Ж.Д. Кардиоренальный синдром (почечный фактор и повышение риска
сердечно-сосудистых заболеваний). Клин фармакол тер 2002; 11: 3: 16-18.
. Мухин
Н.А., Моисеев В.С. Кардиоренальные соотношения и риск сердечно-сосудистых
заболеваний // Вестник РАМН. 2003: 11; 50-55
. Насонов
Е.Л. Антифосфолипидный синдром. - М.: Литтерра, 2004.
. Насонов
Е.Л. ред. Клинические рекомендации. Ревматология. - М.: ГЭОТАР-Медиа, 2008
. Насонова
В.А., Фоломеева О.М., Эрдес Ш.Ф. Ревматические болезни в России в начале XXI
века. Научно-практ ревматол 2003; 1: 6-10.
. Насонов
Е.Л. Кардиоваскулярные эффекты нестероидных противовоспалительных препаратов.
Научно-практ ревматол 2003; 3: 28-31.
. Насонов
Е.Л. Перспективы фармакотерапии воспалительных ревматических заболеваний:
моноклональные антитела к фактору некроза опухоли-альфа. Рус мед журн 2001; 9:
7-8: 280-284.
. Насонов
Е.Л. Моноклональные антитела к фактору некроза опухоли-альфа в ревматологии.
Рус мед журн 2003; 11: 7: 390-394.
. Насонов
Е.Л. Ревматоидный артрит как общемедицинская проблема. Тер арх 2004; 5: 5-7.
. Насонов
Е.Л. Патогенетическое и клиническое обоснование применения статинов при
системной красной волчанке и антифосфолипидном синдроме. Клин фармакол тер
2004; 13: 1: 82-89.
. Насонов
Е.Л. Проблема атеротромбоза в ревматологии. Вестн РАМН 2003; 7: 6-10.
. Оганов
Р.Г., Масленникова Г.Я. Вклад сердечно-сосудистых заболеваний в здоровье
населения России. Сердце 2003; 2: 58-61.
. Палеев
Н.Р., Палеев Ф.Н. Цитокины и их роль в патогенезе заболеваний сердца. Клин мед
2004; 5: 4-7.
. Ребров
А.П., Инамова О.В. Предпосылки развития эндотелиальной дисфункции при
ревматоидном артрите. Тер арх 2004; 5: 79-85.
. Ситникова
М.Ю., Максимова Т.А., Вахрамеева Н.В. и др. Состояние эндотелия и маркеры
хронического воспаления у больных ИБС, осложненной сердечной недостаточностью.
Сердечная недостаточность 2002; 3: 2: 80-82.
. Сперанский
А.И., Иванова С.М. Аутоиммунные болезни и синдромы. Научно-практ. ревматол
2002; 4: 127.
. Старовойтова
М.Н., Гусева Н.Г. Анализ причин смерти больных системной склеродермией.
Научно-практ ревматол 2001; 3: 110.
. Тарасова
И.А., Иванова М.М., Жорняк А.П., Насонова В.А. Значение индекса повреждения в
прогнозировании исхода системной красной волчанки. Тер арх 2003; 1: 59-62.
. Фоломеева
О.М., Тарасова И.А., Дубинина Т.В., Эрдес Ш. Заболеваемость населения России
ревматическими болезнями в 2001-2002 гг. Научно-практ ревматол 2004; 2: 4-7.
. Чазов
Е.И. К вопросу об атеротромботической болезни. Кардиология 2001; 4: 4-7.
. Чазов
Е.И. Взгляд из прошлого в будущее. Тер арх 2004; 6: 8-15.
. Шилкина
Н.П. Васкулиты и васкулопатии. Ярославль: Рико Пресс; 2001.
31. Aringer
M., Smolen J.complex cytokine effect in a complex autoimmune disease, tumor
necrosis factor in systemic lupus erythematosus. Arthr Res Ther 2003; 5:
172-177.
. Aronov
C., Ginzler E. Epidemiology of cardiovascular disease in systemic lupus
erythematosus. Lupus 2000; 9: 166-169.
. Bastos
C. J., Queiroz A. C., Martinelli R. Cardiac involvement in systemic lupus
erythematosus: anatomo-pathological study. Rev Ass Med Bras 1993; 39: 3:
161-164.
. Bruce
I., Gladman D., Urowitz M. Premature atherosclerosis in systemic lupus
erythematosus. Rheum Dis Clin North Am 2000; 26: 257-278.
. Callahan
L. F., Pincus T. Mortality in the rheumatic diseases. Arthr Care Res 1995; 8:
4: 229-241.
36. Cimaz
R, Meroni PL, Shoenfeld Y. Epilepsy as part of systemic lupus erythematosus and systemic
antiphospholipid syndrome (Hughes syndrome). Lupus. 2006; 15 (4): 191-7.
37. Cines
D. B., Pollak E. S., Buck C. A. et al. Endothelial cells in physiology and in
the pathophysiology of vascular disorders. Blood 1998; 91: 10: 3527-3561.
39. Dayer
J. - M., Fenner H. Cytokines and their inhibitors in arthritis. Bailliere Clin
Rheum 1992; 6: 485-516.
. De
Inocencio J., Lovell D. J. Cardiac function in systemic lupus erythematosus. J
Rheumatol 1994; 21: 11: 2147-2156.
. Feldmann
M., Brennan F. M., Maini R. Cytokines in autoimmune disorders. Int Rev Immunol
1998; 17: 217-228.
42. Ha
C, Magowan S, Accortt NA, Chen J, Stone CD. Risk of arterial thrombotic events in
inflammatory bowel disease. Am J Gastroenterol. 2009 Jun; 104 (6): 1445-51.
43. Hahn
B. Systemic lupus erythematosus and accelerated atherosclerosis. New Engl J Med
2003; 349: 2379-2380.
. Hansen
G. Immune mechanisms in atherosclerosis. Atrerioscler Thromb Vasc Biol 2001;
21: 1876-1890.
. Jimenez
S., Ramos-Cassals M., Cevera К. et al. Atherosclerosis in systemic lupus
erythematosus, clinical relevance. In: Atherosclerosis and autoimmunity. Eds.
Y. Schoenfeld, D. Harats, G. Wick. Elsevier 2001; 255-265.
. Manzi
S. Systemic lupus erythematosus, a model for atherogenesis? Rheumatology 2000;
39: 353-359.
. Manzi
S., Meilachn E., Rairi J. et al. Age specific incidence rates of myocardial
infarction and angina in women with systemic lupus erythematosus, comparison
with the Fremingham study. Am J Epidemiol 1997; 145: 408-415.
. Mayet
W. - J., Schwarting A., Orth Th., Meyer zum Buchenfelde K. - H. Cytotoxic
effects of antibodies to proteinase 3 (c-ANCA) on human endothelial cells. Clin
Exp Immunol 1994; 97: 458-465.
49. Miranda CH
<http://www.ncbi.nlm.nih.gov/pubmed?term=Miranda%20CH%5BAuthor%5D&cauthor=true&cauthor_uid=22735914>,
Gali LG
<http://www.ncbi.nlm.nih.gov/pubmed?term=Gali%20LG%5BAuthor%5D&cauthor=true&cauthor_uid=22735914>,
Marin-Neto JA <http://www.ncbi.nlm.nih.gov/pubmed?term=Marin-Neto%20JA%5BAuthor%5D&cauthor=true&cauthor_uid=22735914>,
Louzada-Jъnior P
<http://www.ncbi.nlm.nih.gov/pubmed?term=Louzada-J%C3%BAnior%20P%5BAuthor%5D&cauthor=true&cauthor_uid=22735914>,
Pazin-Filho A
<http://www.ncbi.nlm.nih.gov/pubmed?term=Pazin-Filho%20A%5BAuthor%5D&cauthor=true&cauthor_uid=22735914>.
Coronary thrombosis as the first complication of antiphospholipid syndrome. Arq
Bras Cardiol. <http://www.ncbi.nlm.nih.gov/pubmed/22735914> 2012 Apr; 98 (4): e66-9.
50. Moder K.
G., Miller T. D., Tazelaar H. D. Cardiac involvement in systemic lupus
erythematosus. Mayo Clin Proc 1999; 74: 3: 275-284.
. Roach RE,
Lijfering WM, Flinterman LE, Rosendaal FR, Cannegieter SC. The increased risk
of arterial cardiovascular disease after venous thrombosis is determined by
common etiologic factors. Blood. 2013 May
3.
52. Robinson K.
Risk factor modification for cardiac disease. Med Clin N Am 2000; 84: 1.
. Salmon
J., Rоman M. Accelerated atherosclerosis in systemic lupus
erythematosus, implications for patients manageent. Curr Opin Rheumatol 2001;
13: 341-344.
. Schachimger
V., Btitten M., Zeiher A. Prognostic impact of coronary vasadilator dysfunction
on adverse long-term outcome of coronary heart disease. Circulation 2000; 101:
1899-1906.
. Schwartz
R. S., Datta S. K. Autoimmunity and autoimmune disease. In: Fundamental
Immunology. Ed. W. E. Paul. New York: Raven Press 1989; 819-866.
56. Sharma
AK, Baig MW, Heist EK. Intracardiac
thrombosis and acute myocardial infarction as initial presentation of
antiphospholipid syndrome. Am J Med
Sci. 2011 Sep; 342 (3): 254-6.
57. Sondheimer
H. M., Lorts A. Cardiac involvement in inflammatory disease: systemic lupus
erythematosus, rheumatic fever and Kawasaki disease. Adolesc Med 2001; 12: 1:
69-78.
. Trager
J., Ward M. Mortality and cause of death in systemic lupus erythematosus. Curr
Opin Rheumatol 2001; 13: 345-351.
. Urowitz
M., Gladman D., Bruce I. Atherosclerosis and systemic lupus erythematosus. Curr
Opin Rheumatol 2000; 2: 19-23.
. Van
Doornum S., McColl G., Wick I. P. Accelerated atherosclerosis. An
Extraarticular feature of rheumatoid arthritis. Arthr Rheum 2002; 46: 862-873.
. Wajed J.,
Ahmad Y., Duttington P., Bruce L. Prevention of cardiovascular disease in
systemic lupus erythematosus - proposed guidelines for risk factor management.
Rheumatology 2004; 43: 7-12.
. Atochin
DN, Huang PL. Endothelial nitric oxide synthase transgenic models of
endothelial dysfunction // Pflugers Arch. - 2010; Vol.460, №6. - P.965-74
. Cervera
R, et al. Task Force on Catastrophic Antiphospholipid Syndrome (APS) and
Non-criteria APS Manifestations (II): thrombocytopenia and skin manifestations.
// Lupus. - 2011; Vol. 20, №2. - P.174-81.
. Charakida
M, Tousoulis D, Stefanadis C, Toutouzas P. The impact of platelet glycoprotein
IIIa and Ia polymorphisms in cardiovascular thrombotic disease. // - 2003;
Vol.4, №1. - P.17-22.
. Chen YL
et al. Association of 4G/5G polymorphism in PAI1 promoter with PAI1 level in
deep vein thrombosis. // Zhonghua Yi Xue Yi Chuan Xue Za Zhi. - 2005; Vol.22,
№6. - P.624-7.
. Devreese
K, Hoylaerts MF. Challenges in the diagnosis of the antiphospholipid syndrome.
// Clin Chem. - 2010; Vol.50, №6. - P.503-9.
. Furie B.
Pathogenesis of thrombosis. // Hematology Am Soc Hematol Educ Program. - 2009,
P.255-8
. Gigante A
et al. Antiphospholipid antibodies and renal involvement. // Am J Nephrol. -
2009. - Vol.37, №11. - P.141-6
. Hahn B.
H., McMahon M. A., Wilkinson A. [et al.] /American College of Rheumatology
Guidelines for Screening, Treatment,and Management of Lupus Nephritis //
Arthritis care&Research. - 2012. - Vol.64. - N.6. - P.797-808
. Hunt BJ,
Wu XX, de Laat B, Arslan AA, Stuart-Smith S, Rand JH. Resistance to annexin A5
anticoagulant activity in women with histories for obstetric antiphospholipid
syndrome. // Am J Obstet Gynecol. - 2010. - Vol.24, №7. - P.56-61
. Kim H,
Cho C, Cho Y, Cho S, Yoon K, Kim K. Significant associations of PAI-1 genetic
polymorphisms with osteonecrosis of the femoral head. BMC // Musculoskelet
Disord. - 2011. - Vol.14, №12. - P.160-7
72. Komáromi I, Bagoly Z, Muszbek L. Factor XIII: novel
structural and functional aspects. // J Thromb Haemost. - 2011. - Vol.9, №1. -
P.9-20
. Krone KA,
Allen KL, McCrae KR. Impaired fibrinolysis in the antiphospholipid syndrome. //
Curr Rheumatol Rep. - 2010. - Vol.2, №1. - P.53-7
. Meroni
PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome:
understanding the antibodies. // Nat Rev Rheumatol. - 2011. - Vol.7, №6. -
P.330-8
. Moore C,
Tymvios C, Emerson M. Functional regulation of vascular and platelet activity
during thrombosis by nitric oxide and endothelial nitric oxide synthase. //
Thromb Haemost. - 2010. - Vol.41, №3. - P.141-6
. Moreno
MU, Zalba G. CYBA gene variants as biomarkers for coronary artery disease. //
Drug News Perspect. - 2010. - Vol.23, №5. - P.316-24
. Moulis G,
Delavigne K, Huguet F, Fortenfant F, Beyne-Rauzy O, Adoue D. Antiphospholipid
antibodies and the risk of thrombosis: A comparative survey between chronic
immune thrombocytopenia and primary antiphospholipid syndrome. // Rev Med
Interne. - 2011. - Vol.16, №10. - P.41-5
. Nalli C
et al. Fine specificity of anti-β2glycoprotein I antibodies in systemic autoimmune
diseases is mostly directed against domain 1 // Reumatismo. - 2011. - Vol.63,
№2. - P.91-6
. Passam
FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular pathophysiology of
the antiphospholipid syndrome: the role of oxidative post-translational
modification of beta 2 glycoprotein I. // J Thromb Haemost. - 2011. - Vol.9,
№1. - P.242-7
. Pastinen
T et al. Array-based multiplex analysis of candidate genes reveals two
independent and additive genetic risk factors for myocardial infarction in the
Finnish population. // Hum Mol Genet. - 1998. - Vol.7, №9. - P.1453-9
. Plowman
EK, Okun MS. Antiphospholipid syndrome and other lupus-related movement
disorders. // Handb Clin Neurol. - 2011. - Vol.4, №11. - P.414-25
. Ramesh S
et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion
and thrombosisin mice by antagonizing eNOS via β2GPI and apoER2. // J Clin Invest. - 2011. - Vol.121,
№1. - P.120-5.
. Rebrov A.
P., Oxenchuk A. N., Karpova O. G. / OCCURRENCE AND STRUCTURE OF CHRONIC KIDNEY
DISEASE IN SYSTEMIC LUPUS ERYTHEMATOSUS // УДК 616.5-002.525.2-06:
616.61-036.12-039.4/.5 (045), 2013
. Rodriguez-Garcia
JL, Bertolaccini ML, Cuadrado MJ, Sanna G, Ateka-Barrutia O, Khamashta MA.
Clinical manifestations of antiphospholipid syndrome (APS) with and without
antiphospholipid antibodies (the so-called 'seronegative APS'). // Ann Rheum
Dis. - 2011. - Vol.105, №5. - P.431-9
. Ronco C.,
Haapio M., House A. et al. Cardiorenal
syndrome // JACC. 2008: 52 (19); 1527-1539;
86. Sangle NA,
Smock KJ. Antiphospholipid antibody syndrome. // Arch Pathol Lab Med. - 2011. -
Vol.135, №9. - P.1092-9.
. Tincani
A, Casu C, Cartella S, Ziglioli T, Cattaneo R. Antiphospholipid antibody:
laboratory, pathogenesis and clinical manifestations // Reumatismo. - 2010. -
Vol.62, №1. - P.65-71
. Tripodi
A, de Groot PG, Pengo V. Antiphospholipid syndrome: laboratory detection,
mechanisms of action and treatment. // J Intern Med. - 2011. - Vol.270, №2. -
P.678-72