Атомная спектроскопия и потенциометрия
Министерство сельского хозяйства Российской Федерации
ФГБОУ ВПО «ВЕЛИКОЛУКСКАЯ ГСХА»
Кафедра химии, агрохимии и агроэкологии
Автореферат
по
дисциплине «Физико-химические методы анализа»
на тему:
«Атомная спектроскопия и потенциометрия»
Великие
Луки 2014
ВВЕДЕНИЕ
Методы атомной спектроскопии - используются для
массовых, быстрых, селективных и достаточно точных определений малых содержаний
элементов.
Для одновременного определения нескольких
элементов, а также качественного анализа наилучшими являются методы
атомно-эмиссионной и рентгенофлуоресцентной спектроскопии.
Использование различных источников атомизации
позволяет определять методом атомно-эмиссионной спектроскопии как основные, так
и примесные компоненты, анализировать растворы и твёрдые образцы. Важнейшим
достоинством рентгенофлуоресцентной спектроскопии является то, что это
неразрушающий метод анализа, что очень важно, например, при анализе
произведений искусства, археологических объектов и т.д. Методы
рентгеноэмиссионной, фото- и оже-электронной спектроскопии - используются для
локального анализа и анализа поверхности твердых тел.
Широкое применение также нашел метод
атомно-абсорбционной спектроскопии. Этим методом можно определять около 6 - 70
элементов, главным образом металлов, при очень малых их концентрациях. Однако
атомно-абсорбционную спектроскопию целесообразно применять лишь для
одноэлементных анализов. От других физико-химических методов ионометрия (прямая
потенциометрия) отличается простотой методик и дешевизной измерительных
приборов. Современные портативные ионометры позволяют определять разнообразные
ионы и растворенные газы не только в лаборатории, но и в полевых условиях.
Сферы применения методов очень многообразны. Это анализ объектов окружающей
среды, продуктов питания, лекарственных препаратов, продукции металлургической,
строительной, стекольной промышленности, геологических образцов. В том числе
атомную спектроскопию применяют в полевых условиях (с использованием
портативных рентгенофлуоресцентных спектрометров).
1.
Атомная спектроскопия
.1. Общие положения
Методы атомной спектроскопии основаны на
переходах валентных (рис. 1.1, a-в) или внутренних (рис. 1.1, г-ж) электронов
атомов из одного состояния в другое.
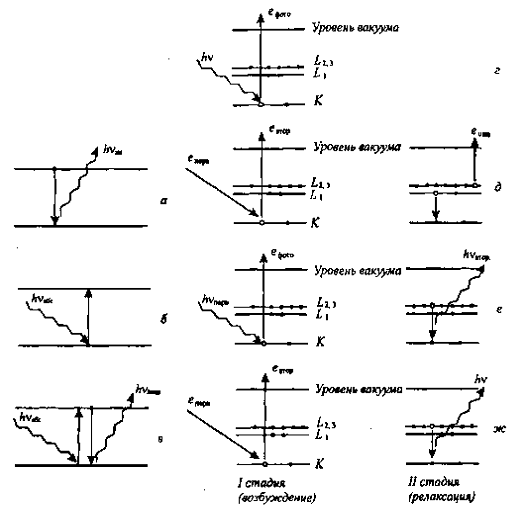
Рис 1.1. Схемы процессов, лежащих в основе
методов спектроскопии: а атомно-эмиссионной; б-атомно-абсорбционной; в -
атомно-флуоресцентной; г - рентгеновской фотоэлектронной; д - оже-электронной;
е -peнтгeнофлуоресцентного анализа; ж -рентгеноэмиссионного анализа. Уровни
энергии электронов: а-б -валентные; г-ж -внутренние испусканием атомом одного
или нескольких электронов (ионизацией)
Поэтому в методах атомной спектроскопии возможна
регистрация как электромагнитных, так и электронных спектров - распределений,
соответственно, фотонов и испускаемых электронов по их энергиям.
Одна из особенностей атомных спектров - их
линейчатая структура. Положения линий индивидуальны для каждого элемента и
могут быть использованы для качественного анализа. На зависимости интенсивности
спектральной линии основан количественный анализ. Относительно мала вероятность
наложения линий различных элементов. Поэтому многие методы атомной
спектроскопии можно использовать для обнаружения и определения одновременно
нескольких элементов.
В зависимости от используемого диапазона длин
волн электромагнитного излучения и природы соответствующих электронных
переходов методы атомной спектроскопии делятся на оптические и рентгеновские.
В методах оптической спектроскопии используют
излучение видимой и УФ-областей оптического диапазона. Оно соответствует
изменению энергии валентных электронов. Для получения оптических атомных
спектров необходима предварительная атомизация пробы - перевод ее в
газообразное атомарное состояние. Для этой цели служат атомизаторы - источники
высокой температуры различной конструкции. Взаимодействие вещества с излучением
оптического диапазона, как правило, не сопровождается ионизацией атомов.
Поэтому для оптического диапазона характерны только методы спектроскопии
электромагнитного излучения. К ним относятся методы атомно-эмиссионной (АЭС),
атомно-флуоресцентной (АФС), атомно-абсорбционной (ААС) спектроскопии.
В методах рентгеновской спектроскопии используют
излучение рентгеновского диапазона, соответствующее изменению энергии внутренних
электронов. Структуры энергетических уровней внутренних электронов в атомарном
и молекулярном состояниях очень близки. Поэтому в рентгеновских методах
атомизация пробы не требуется.
Однако взаимодействие вещества с излучением
рентгеновского диапазона всегда сопровождается ионизацией атомов.
Такая ионизация происходит под действием
внешнего источника рентгеновского излучения или пучка высокоэнергетических
электронов. Электрон, испускаемый атомом вследствие ионизации, называют
фотоэлектроном или, соответственно, вторичным электроном. В результате
внутриатомных электронных переходов возможна эмиссия еще одного электрона,
называемого оже-электроном. При использовании рентгеновского излучения возможна
регистрация как электромагнитных, так и электронных спектров.
К рентгеновским методам спектроскопии
электромагнитного излучения относят рентгеноэмиссионный анализ (РЭА),
рентгенофлуоресцентный (РФА) и рентгеноабсорбционный (РАА) анализ, а к методам
электронной спектроскопии рентгеновскую фотоэлектронную (РФЭС) и оже-электронную
(ОЭС) спектроскопию.
В зависимости от физической природы процесса
взаимодействия излучения с веществом методы атомной спектроскопии
электромагнитного излучения (как оптического, так и рентгеновского диапазона)
делят на эмиссионные и абсорбционные. В оптических эмиссионных методах для
получения спектра испускания необходим предварительный перевод атомов в
возбужденное состояние. Для этой цели служат устройства, называемые источниками
возбуждения, источники высокой температуры (для оптических методов), потоки
высокоэнергетических частиц (для рентгеновских методов), электромагнитное
излучение. Эмиссионные оптические методы, в которых возбуждение атомов
происходит под действием высокой температуры, называют методами
атомно-эмиссионной спектроскопии (АЭС). В этих методах атомизатор - источник
возбуждения. Если источником возбуждения служит электромагнитное излучение,
методы называют флуоресцентными - атомно-флуоресцентная спектроскопия (АФС),
рентгенофлуоресцентный анализ (РФА).
В абсорбционных методах возбуждение атомов не
требуется, источники возбуждения отсутствуют.
Классификация основных методов атомной
спектроскопии приведена в табл. 1
Таблица 1. Классификация основных методов
спектроскопии
|
Метод
|
Диапазон
электромаг. излучения
|
Процесс
|
Способ
|
|
|
|
атомизации
|
возбуждения
|
регистрации
|
|
Атомно-эмиссионный
(АЭС)
|
Оптический
|
Эмиссия
фотонов
|
Высоко-температурный
|
Высоко-температурный
|
Электромагнитная
|
|
Атомно-
флуоресцентный (АФС)
|
|
|
|
Электромагнитное
излучение (УФ-видимое)
|
|
|
Атомно-абсорбционный
(ААС)
|
|
Абсорбция
фотонов
|
|
Не
требуется
|
|
|
Рентгено-эмиссионный
(РЭА)
|
Рентгеновский
|
Эмиссия
фотонов
|
Не
требуется
|
Поток
электронов
|
|
|
Рентгено-флуоресцентный
(РФА)
|
|
|
|
Электромагнитное
излучение (рентген.)
|
|
|
Рентгено-абсорбционный
(РАА)
|
|
Абсорбция
фотонов
|
|
Не
требуется
|
|
|
Рентгеновский
фотоэлектронный (РФЭС)
|
Регистрация
электронного спектра с кинетической энегрией до 1500 эВ
|
Эмиссия
электронов
|
|
Электромагнитное
излучение (рентген.)
|
Электронная
|
|
Оже-электронный
(ОЭС)
|
|
|
|
Поток
электронов
|
|
1.2. Атомно-эмиссионная
спектроскопия
.2.1. Основы метода
Метод атомно-эмиссионной спектроскопии (АЭС)
основан на термическом возбуждении свободных атомов или одноатомных ионов и
регистрации оптического спектра испускания возбужденных атомов (см. рис. 1.1,
а). Аналитическим сигналом в АЭС служит интенсивность испускаемого излучения I.
Поскольку возбуждение атомов имеет термическую природу, возбужденные и
невозбужденные атомы находятся между собой в термодинамическом равновесии,
положение которого описывается законом распределения Больцмана
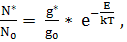 (1.1)
(1.1)
где No - число невозбужденных атомов; g* и go
- статистические веса возбужденного и невозбужденного состояния; Е - энергия
возбуждения; k - постоянная Больцмана; Т - абсолютная температура. Таким
образом, при постоянной температуре число возбужденных частиц N* прямо
пропорционально числу невозбужденных частиц No , т. е. фактически общему числу
данных атомов N в атомизаторе. В свою очередь (при заданных условиях
атомизации, определяемых конструкцией и режимом работы прибора и рядом других
факторов), число атомов (N)
в атомизаторе пропорционально концентрации определяемого элемента в пробе С.
Таким образом, можно было бы ожидать, что между интенсивностью испускаемого
излучения I и концентрацией
определяемого элемента С наблюдается прямо пропорциональная зависимость. Однако
на практике условия, обеспечивающие эту зависимость, выполняются далеко не
всегда. В общем случае зависимость интенсивности излучения от концентрации
нелинейна и может быть описана эмпирическим уравнением
 (1.2)
(1.2)
Коэффициент а в уравнении (1.2) является сугубо
эмпирической величиной, зависящей от условий процесса. Поэтому в АЭС решающее
значение имеет правильный выбор условий атомизации и измерения аналитического
сигнала, включая градуировку по образцам сравнения.
1.2.2. Атомизаторы
Основные типы источников атомизации и возбуждения,
применяемых в АЭС, приведены в табл. 2.
Таблица 2. Основные типы атомизаторов в АЭС
|
Тип
источника атомизации
|
Т,
°С
|
Состояние
пробы
|
Смin
, % масс.
|
Sг
|
|
Пламя
|
1500
- 1300
|
Раствор
|
10-7
- 10-2
|
0,01
- 0,5
|
|
Электрическая
дуга
|
3000
- 7000
|
Твердая
|
10-4
- 10-2
|
0,1
- 0,2
|
|
Электрическая
искра
|
~10000
- 12000
|
Твердая
|
10-3
- 10-1
|
0,05
- 0,1
|
|
Индуктивно
связанная плазма (ИСП)
|
6000
- 10000
|
Раствор
|
10-3
- 10-2
|
0,01
- 0,05
|
Важнейшей характеристикой любого атомизатора
является его температура атомизации. От нее зависит физико-химическое состояние
анализируемого вещества, величина аналитического сигнала и метрологические
характеристики методики. Как видно из табл. 2, атомизаторы, используемые в АЭС,
значительно различаются по своей температуре.
Пламя. Вариант АЭС с атомизацией в пламени
называют методом эмиссионной фотометрии пламени. Конструктивно пламенный
атомизатор для АЭС представляет собой горелку (рис. 1.2).
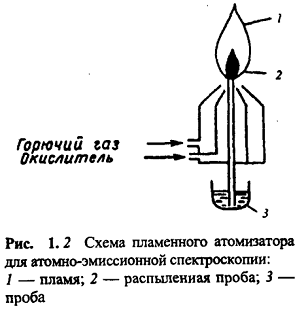
Анализируемую пробу (раствор) распыляют с
помощью форсунки в пламя. Пламя состоит из двух основных зон: восстановительной
и окислительной. В восстановительной зоне протекают первичные реакции
термической диссоциации и неполного сгорания компонентов горючей смеси. Эта
зона, а также внутренний конус, находящийся между восстановительной зоной и
окислительной, интенсивно излучают свет почти во всем УФ-видимом диапазоне. Это
излучение накладывается на линии испускания возбужденных атомов и поэтому
восстановительную зону пламени для аналитических целей не используют. В
окислительной зоне пламени происходят реакции полного сгорания компонентов
смеси с образованием Н2O и CO2. Эта зона интенсивно излучает в ИК-области и
мало в УФ- и видимой областях, поэтому ее используют для аналитических целей.
Температуру, состав и окислительно-восстановительные свойства пламени можно
регулировать, варьируя соотношением горючего газа и окислителя в смеси. Этот
прием часто используют для выбора оптимальных условий атомизации и устранения
физико-химических помех.

В зависимости от состава горючей смеси
температура пламени может составлять от 1500 (светильный газ - воздух) до 3000
°С (С2Н2 - N20). Эти температуры оптимальны для определения лишь наиболее легко
атомизируемых и возбудимых элементов, в первую очередь щелочных и
щелочно-земельных (Са, Sг, Ва) металлов. Для них метод фотометрии пламени
является одним из самых чувствительных (пределы обнаружения до 10-7 % масс.).
Для большинства других элементов пределы обнаружения на несколько порядков
выше. Важное достоинство пламени как источника атомизации - высокая
стабильность и связанная с ней хорошая воспроизводимость результатов измерений
(Sг= 0,0 1 - 0,05).
Электрическая дуга. В АЭС используют дуговые
разряды постоянного и переменного тока. Дуговой атомизатор представляет собой
пару электродов (чаще всего угольных), между которыми пропускают электрический
разряд. Нижний электрод имеет углубление, в которое помещают пробу. Дуговой
разряд наиболее удобен для анализа твердых проб. Для анализа растворов пробу
выпаривают вместе с инертным порошкообразным материалом (коллектором), а затем
помещают в углубление электрода. Если анализируемая проба - металл (сплав), то
она служит нижним электродом.
Температура дугового разряда существенно выше:
3000 - 7000 °С. Таких температур вполне достаточно для эффективной атомизации и
возбуждения большинства элементов (кроме галогенов). Поэтому для большинства
элементов пределы обнаружения в дуговом разряде в среднем составляют 10-4 -
10-2 % масс. Для дуги переменного тока температура несколько выше, чем для дуги
постоянного тока. Дуговые атомизаторы (особенно постоянного тока) не отличаются
высокой стабильностью режима работы. Поэтому воспроизводимость результатов
невелика: Sг = 0,1 - 0,2. Однако для полуколичественных определений такая
воспроизводимость вполне достаточна. Одна из наиболее важных областей
применения дуговых атомизаторов - это качественный анализ на основе обзорного
спектра.
Электрическая искра. Искровой атомизатор устроен
так же, как и дуговой. В спектральных приборах для генерации дугового и
искрового разрядов используют одно и то же устройство, а выбор типа разряда
осуществляется переключением электрической схемы. Искровой атомизатор
предназначен для анализа твердых образцов (иногда вводят жидкие пробы в виде аэрозоля
в разрядный промежуток между электродами). Особенность искрового атомизатора -
отсутствие термодинамического равновесия между находящимися в нем частицами.
Поэтому говорить о температуре искрового разряда достаточно сложно. Его
эффективная температура достигает около 10000 °С. Этого достаточно для
возбуждения даже наиболее трудновозбудимых галогенов. Искровой разряд
стабильнее дугового, и воспроизводимость результатов выше (Sг = 0,05 - 0,1).
Индуктивно связанная плазма. Это самый
современный источник атомизации, обладающий наилучшими аналитическими
возможностями и метрологическими характеристиками. Атомизатор с ИСП
представляет собой плазменную горелку особой конструкции (рис. 1.4), состоящую
из трех концентрических кварцевых трубок. В них с большой скоростью подают
потоки особо чистого аргона. Самый внутренний поток служит для впрыскивания
раствора пробы, средний является плазмообразующим, а внешний служит для
охлаждения плазмы; расход аргона в этом потоке особенно велик (1-20 л/мин).
Аргоновая плазма инициируется (поджигается)
искровым разрядом, а затем стабилизируется с помощью высокочастотной
индуктивной катушки, окружающей верхнюю часть горелки. При этом возникает
сильный кольцевой ток заряженных частиц, находящихся в плазме. Температура
аргоновой плазмы изменяется по высоте горелки и составляет 60 - 10000 °С.

Метод ИСП-АЭС характеризуется универсальностью
(при столь высоких температурах возбуждается большинство элементов), высокой
чувствительностью (Cмin
= 1 0-8 - 1 0-2 % масс.), хорошей воспроизводимостью (Sг = 0,01 - 0,05) и
широким диапазоном определяемых концентраций. Основной фактор, сдерживающий
применение ИСП в аналитической практике - высокая стоимость оборудования и
расходуемых материалов (аргона высокой чистоты).
1.2.3. Спектральные
помехи
Зависимость между интенсивностью испускания и
концентрацией определяемого элемента в пробе является нелинейной [см. уравнение
(1.2)]. Это обусловлено разными причинами: одни из них связаны с
взаимодействием вещества с излучением (спектральные помехи), другие - с
взаимодействием веществ между собой, низкой эффективностью распыления, неполным
испарением пробы и другими физическими и химическими причинами
(физико-химические помехи).
Самопоглощение - явление, которое в той или иной
мере наблюдается в любых эмиссионных методах анализа. Сущность: часть излучения
возбужденных атомов может поглотиться невозбужденными атомами того же элемента,
находящимися в периферийной части атомизатора. В результате регистрируемая
интенсивность уменьшится. Поскольку в периферийной части атомизатора
температура ниже, чем в центральной, то ширина атомной линии поглощения меньше,
чем ширина линии испускания. Поэтому наиболее интенсивно будет поглощаться
часть излучения вблизи максимума линии. Это может привести к самообращению
линии испускания - ее кажущемуся расщеплению на две линии (рис. 1.5).

Степень самопоглощения возрастает с увеличением
концентрации атомного пара. Это приводит к нарушению линейной зависимости I
от С в области высоких концентраций (рис. 1.6, а). Самопоглощение - главная
причина, обусловливающая нелинейный характер зависимости интенсивности
излучения от концентрации элемента, описываемой уравнением Ломакина-Шайбе
(1.2).
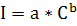 (1.3)
(1.3)
где b
- параметр, характеризующий степень самопоглощения, является функцией
концентрации и при ее увеличении непрерывно изменяется от 1(отсутствие
поглощения) до 0. Однако при работе в достаточно узком концентрационном
диапазоне величину b можно
считать практически постоянной. В этом случае зависимость интенсивности от
концентрации в билогарифмических координатах близка к прямолинейной даже в том
случае, когда b < 1 (рис. 1.6,
б).
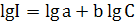 (1.4)
(1.4)
Переход к билогaрифмическим координатам
позволяет использовать для аналитических целей и нелинейный участок зависимости
I от С.
Степень поглощения во многом определяется
геометрией атомизатора. Для источников ИСП самопоглощение значительно ниже, чем
для других атомизатров.

Излучение и поглощение фона. Наряду со
свободными атомами в атомизаторе присутствуют и многоатомные частицы -
молекулы, свободные радикалы. При высокой температуре они могут возбуждаться и
испускать излучение в оптическом диапазоне. Это излучение (а также излучение
раскаленных макроскопических частиц) называется фоновым. В частности, при
использовании дуговой или искровой атомизации с угольными электродами возникает
интенсивное фоновое излучение молекул CN в области 360 - 460 нм. При
возникновении интенсивных фоновых помех использовать соответствующую область
спектра в аналитических целях невозможно. Если же излучение фона не очень
велико, его влияние можно скомпенсировать. Для этого измеряют интенсивность
фонового излучения при длине волны в непосредственной близости от изучаемой
спектральной линии и вычитают ее из интенсивности спектральной линии. Мешающее
влияние оказывает, и поглощение фоном части излучения, испускаемого
возбужденными атомами.
Наложение атомных спектральных линий. Спектр
испускания каждого элемента состоит из большого числа линий, отвечающих
различным переходам из возбужденных состояний в состояния с меньшей энергией.
Число таких линий может измеряться тысячами. Поэтому в АЭС высока вероятность
наложения спектральных линий различных элементов друг на друга. В этих случаях
для анализа необходимо использовать линии спектра, свободные от наложений. При
повышении температуры атомные эмиссионные спектры усложняются. Расшифровку
спектров, полученных при высоких температурах (ИСП, искра), часто необходимо
проводить компьютерными методами.
С метрологической точки зрения спектральные
помехи многих типов, например, наложение спектральных линий, имеют аддитивный
характер, т. е. вклады в общий аналитический сигнал, вносимые определяемым и
посторонним компонентами, суммируются. Это обстоятельство необходимо учитывать
при выборе способа градуировки в количественном анализе.
1.2.4. Физико-химические
помехи
Общую схему процессов, происходящих с веществом
в атомизаторе при высокой температуре, можно представить следующим образом:
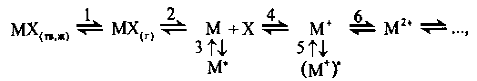 (1.5)
(1.5)
где М - определяемый элемент; М*-
возбужденное состояние.
В атомизаторе проба, исходно находящаяся в
твердом или жидком состоянии, испаряется (стадия 1), затем атомизируется
(стадия 2). Свободные атомы М участвуют далее в двух независимых параллельных
процессах: возбуждении (стадия 3) и ионизации (стадия 4); образующиеся при этом
ионы М+ также могут возбуждаться (стадия 5) или ионизироваться далее (стадия
6).
Таким образом, вещество в атомизаторе находится
в большом числе форм, из которых аналитический сигнал формируют лишь
возбужденные одноатомные частицы. Любой фактор, снижающий их концентрацию,
приводит к уменьшению аналитического сигнала. Рассмотрим основные физические и
химические факторы, влияющие на концентрацию возбужденных частиц в атомизаторе.
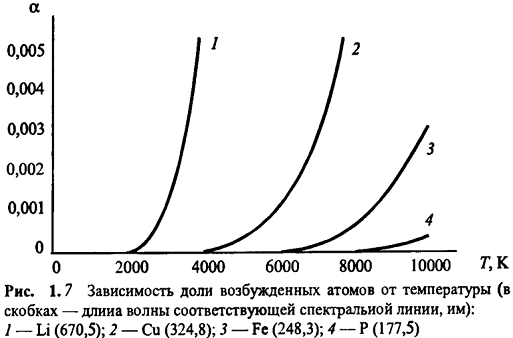
Температура атомизатора непосредственно влияет
на величину аналитического сигнала в АЭС, поскольку от нее, в соответствии с уравнением
Больцмана (1.1), зависит, доля возбужденных частиц. Эта зависимость выражена
достаточно резко (рис. 1.7). Помимо этого, температура влияет на величину
аналитического сигнала и опосредовано, поскольку от нее зависят полнота
атомизации пробы и степень ионизации определяемого элемента.
Полнота испарения и атомизации пробы. Как
испарение [см. уравнение (1.5), стадия 1], так и атомизация (стадия 2) являются
эндотермическими процессами, поэтому их протеканию благоприятствует повышение
температуры. Неполное испарение и атомизация могут серьезно повлиять на
результаты анализа при использовании пламенных атомизаторов. Температура в них
относительно невысока, а проба подается в виде раствора с большой скоростью и
находится в атомизаторе весьма незначительное время. Степень атомизации
вещества в пламени зависит от конструкции атомизатора (в первую очередь -
распыляющей форсунки) и режима его работы, но, как правило, никогда не
превышает нескольких процентов от общего содержания определяемого компонента.
Атомизацию можно увеличить введением в распыляемый раствор специальных добавок
(например, поверхностно-активных веществ), уменьшающих вязкость и поверхностное
натяжение раствора и тем самым способствующих его диспергированию. При
использовании электроразрядных атомизаторов (дуга, искра), предназначенных для
анализа твердых образцов, степень атомизации сильно зависит от физического
состояния пробы. Например, при анализе образцов сплава и минерала с одинаковым
содержанием меди в одинаковых условиях интенсивность испускания линий меди
может быть различной.
Неполнота испарения и атомизации представляет
собой серьезную проблему, если определяемый элемент склонен к образованию
труднолетучих или труднодиссоциирующих (термически устойчивых) соединений. В
этих случаях степень атомизации и интенсивность испускания может сильно
зависеть от валового состава пробы (матричные эффекты) - как катионного, так и
анионного. Например, при прочих равных условиях интенсивность линий испускания
кальция для хлоридных растворов выше, чем для фосфатных, поскольку в последних
кальций образует термически устойчивые фосфаты. Если в растворе, содержащем
кальций, присутствует алюминий, интенсивность испускания атомов кальция также
снижается ввиду образования смешанных оксидов кальция и алюминия.
Ионизация. Процесс ионизации [уравнение (1.5),
стадия 4] конкурирует с процессом возбуждения (стадия 3) и, таким образом,
также снижает величину аналитического сигнала. Этот эффект выражен особенно
сильно для легкоионизирующихся элементов - Na, К, Са и др. Ионизация - тоже
эндотермический процесс, поэтому степень ионизации возрастает при увеличении
температуры.
Физико-химические помехи в АЭС могут вызвать
снижение чувствительности определения, ухудшение правильности и воспроизводимости
результатов, а также нарушение линейности градуировочной зависимости.
Действительно, равновесия атомизации
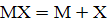
и ионизации
представляют собой равновесия ассоциации-диссоциации,
положение которых зависит от общей концентрации вещества.
При увеличении концентрации возрастает доля
недиссоциированных частиц, при уменьшении - доля ионов. Из-за высокой степени
ионизации в области малых концентраций градуировочные графики в АЭС могут иметь
S-образную форму (см. рис. 1.6, кривая 2).
Основными приемами подавления физико-химических
помех служат изменение температуры атомизатора и использование
спектроскопических буферов. При анализе твердых образцов с использованием
электроразрядных атомизаторов, кроме того, применяют специфические приемы
предварительного удаления мешающих компонентов, называемые обжигом или
обыскриванием.

Температура атомизатора. Увеличение температуры
благоприятствует атомизации и возбуждению атомов, но увеличивает степень их
ионизации. Поэтому для каждого элемента существует своя оптимальная температура
атомизации в АЭС (рис. 1.8). Чем легче атомизируется, возбуждается и
ионизируется определяемый элемент, тем ниже должна быть температура
атомизатора. Так, для определения кальция оптимальная температура атомизации
существенно ниже, чем для определения бора (ср. кривые 1 и 3, рис. 1.8). При
использовании линий испускания возбужденных ионов требуется значительно более
высокая температура, чем в случае нейтральных атомов (ср. кривые 1 и 2, рис
1.8). В ряде случаев подобные температуры для атомизатора данного типа могут
быть технически недостижимы, и определение элемента по линиям ионов становится
невозможным.
Модификаторы матрицы - это вещества, специально
добавляемые к пробе для смещения физико-химических равновесий в газовой фазе,
изменения состава или физических свойств образца либо среды атомизатора,
стабилизации ее параметров с целью увеличения концентрации свободных атомов в
атомизаторе и увеличения аналитического сигнала. В зависимости от механизма
действия модификаторов матрицы (которые очень разнообразны) их называют
буферами, носителями, плавнями, освобождающими добавками и т. д. (общепринятой
классификации и терминологии не существует). Например, для увеличения степени
атомизации трудно диссоцирующих оксидов Si, AI, Ва, Ti
и других к пробе часто добавляют соединения лантана, образующего в газовой фазе
весьма прочные оксиды и тем самым способствующие атомизации элемента М.
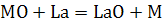
Добавление летучих солей аммония, например
NH4CI, также часто улучшает атомизацию, но уже по совершенно другой причине:
соли аммония, превращаясь при нагревании в газообразные продукты, способствуют
более тонкому диспергированию вещества пробы и тем самым облегчают ее
испарение. Для уменьшения степени ионизации легко ионизирующихся элементов
добавляют соединение элемента (например, калия), ионизирующегося еще легче, чем
определяемый. За счет его ионизации возрастает концентрация электронов в
газовой фазе, и ионизация определяемого элемента М подавляется:
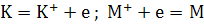
Вещества, стабилизирующие физические параметры
среды атомизатора (температура, вязкость и др.) называют спектроскопическими буферами.
Например, упоминавшиеся соли щелочных металлов играют также роль
спектроскопического буфера, поддерживая на постоянном уровне температуру
дугового или искрового разряда. Дополнительная энергия, подводимая к
атомизатору, в этом случае расходуется не на повышение его температуры, а на
испарение, диссоциацию и ионизацию вещества-буфера.
Обжиг, обыскриваиие. Это специальные приемы
подавления матричных эффектов, широко применяемые при анализе твердых образцов
с использованием электроразрядных атомизаторов. В случае дугового разряда этот
прием называется обжигом, в случае искрового - обыскриванием. Сущность обжига и
обыскривания состоит в предварительном (до регистрации спектра) кратковременном
пропускании соответствующего электрического разряда через анализируемый
образец. При этом из пробы удаляются мешающие компоненты, более летучие, чем
определяемый (например, углерод, фосфор, сера - при анализе сталей). В
результате матричные эффекты существенно уменьшаются, а правильность
результатов улучшается.
Таким образом, степень физико-химических помех в
АЭС зависит от способа и условий атомизации. Источником, наиболее свободным от
физико- химических помех, является ИСП, в которой проба изолирована от
окружающей среды инертным газом - аргоном. С метрологической точки зрения
физико-химические помехи являются мультипликативными (пропорциональными). Это
означает, что мешающие факторы не вносят собственного вклада в величину
аналитического сигнала, однако влияют на величину коэффициента чувствительности
а [см. уравнение (1.3)]. Эффективным приемом подавления такого рода помех
служит метод добавок.
электрод электронный спектроскопия
индикаторный
1.2.5. Метрологические
характеристики и аналитические возможности атомно-эмиссионного метода
Чувствительность. Пределы обнаружения в АЭС
зависят от способа атомизации и природы определяемого элемента и могут
изменяться в широких пределах (см. табл. 1). Для легковозбудимых и
легкоионизирующихся элементов (щелочные и щелочноземельные металлы) наилучшим
источником атомизации является пламя (Cmin до 10-7 % масс.). Для большинства
других элементов наивысшая чувствительность достигается при использовании ИСП
(до 10-8 % масс.). Традиционные источники атомизации - дуга и искра - наименее
чувствительны. Пределы обнаружения в искровом разряде на 1-2 порядка выше, чем
в дуговом из-за того, что он происходит в меньшей области пространства. Это
уменьшает количество испаряемой пробы.
Диапазон определяемых содержаний. Верхние
границы определяемых содержаний в АЭС лимитируются самопоглощением и нарушением
линейности градуировочной характеристики. В зависимости от содержания элемента
для его определения можно использовать линии разной интенсивности. При наличии
самопоглощения можно в достаточно узком интервале концентраций линеаризовать
градуировочную характеристику путем перехода к билогарифмическим координатам
(см. рис. 1.6, б). Так, диапазон определяемых концентраций состоит из
нескольких отдельных поддиапазонов, каждый из которых перекрывает не более 1
порядка, а все вместе - 2-3 порядка. Для ИСП самопоглощение крайне
незначительно, а «единый» диапазон линейности может достигать 4-5 порядков
величин концентрации.
Воспроизводимость. В АЭС аналитический сигнал
пропорционален заселенности возбужденного состояния атомов и весьма
чувствителен к изменениям температуры. Для наиболее стабильных источников
атомизации (пламя, ИСП) величина Sr
= 0,01-0,05, что является хорошей воспроизводимостью для инструментальных
методов анализа. Однако для искрового и особенно дугового разрядов
воспроизводимость существенно хуже (Sг
= 0,05-0,1 и 0,1-0,2, соответственно).
Для улучшения воспроизводимости в АЭС широко
применяют метод внутреннего стандарта. Внутренний стандарт - компонент,
содержание которого во всех образцах, применяемых для градуировки, а также в
анализируемом образце, одинаково. Чаще всего это компонент основы, например,
при анализе сталей внутренний стандарт - железо. При отсутствии подходящего
компонента внутренний стандарт во все образцы вводят специально. Сущность
метода в том, что в качестве аналитического сигнала вместо абсолютной
интенсивности линии определяемого элемента используют отношение I/I0
двух одновременно измеряемых интенсивностей линий - определяемого элемента (I)
и внутреннего стандарта (I0).
Такая пара линий называется гомологической. Если колебания температуры (а также
других условий анализа) влияют на величины I
и I0 в равной степени,
то при вычислении отношения I/I0
эти влияния взаимно компенсируются, и воспроизводимость результатов значительно
улучшается.
Качественный анализ. Атомно-эмиссионный метод
позволяет одновременно зарегистрировать множество линий испускания. Это
позволяет успешно использовать его для качественного анализа. Из традиционных
источников атомизации наиболее подходит дуговой разряд. С одной стороны,
температура дуги достаточна для атомизации и возбуждения большинства элементов.
С другой стороны дуговые спектры существенно беднее линиями, чем искровые и
ИСП, что облегчает идентификацию. Основной недостаток дугового разряда - низкая
стабильность. На качественный анализ не играет существенной роли, поскольку для
идентификации используют положение (длину волны) линии в спектре, а не ее
интенсивность.
Количественный анализ. При количественном
анализе методом АЭС можно использовать все основные способы градуировки -
внешних стандартов (градуировочного графика), внутреннего стандарта и метод
добавок. Целесообразность применения каждого способа зависит от характера
возможных помех и природы анализируемого объекта. Так, метод добавок позволяет
эффективно устранить косвенные мультипликативные погрешности, вызываемые
физико-химическими помехами, однако против наложения спектральных линий - он
бессилен. Следует в то же время иметь в виду, что метод добавок легко реализуем
технически только при анализе растворов (пламя, ИСП). В любом случае при
построении градуировочной зависимости следует стремиться к тому, чтобы все
образцы были максимально адекватны анализируемому - по валовому химическому
составу и физическому состоянию
1.2.6. Способы
монохроматизации и регистрации спектров
Способы разложения излучения в спектр в АЭС
тесно связаны со способами регистрации спектра. Основные способы регистрации-
фотоэлектрический и фотохимический (фотографический). Для массовых
полуколичественных анализов используют приборы с визуальной регистрацией
спектров (стилоскопы). Детекторами для фотоэлектрической регистрации служат
преобразователи: фотоэлементы, фотоэлектронные умножители, фотодиоды.
Фотохимические детекторы - это фотопластинки или фотопленки. В этом случае
интенсивность светового потока определяет величину почернения (оптической
плотности) изображения спектральной линии на пластинке (пленке). Величину
почернения измеряют фотометрическим методом.
В АЭС применяют одно- и многоканальные способы
регистрации спектров. Для разложения излучения пробы в спектр в АЭС используют
моно- и полихроматоры. Как правило, атомно-эмиссионные спектры весьма богаты
линиями, поэтому необходимо использование Моно- и полихроматоров достаточно
высокого разрешения. При пламенной атомизации ввиду малого числа наблюдаемых в
этих условиях эмиссионных линий можно использовать и светофильтры.
1.3. Атомно-абсорбционная
спектроскопия
.3.1. Основы метода
Атомно-абсорбционная спектроскопия (ААС)
основана на поглощении излучения оптического диапазона невозбужденными
свободными атомами (см. рис. 1.1, б). Таким образом, в ААС, как и в АЭС,
необходима предварительная атомизация пробы. Однако если в АЭС аналитический
сигнал формируют возбужденные атомы, то в ААС невозбужденные. Величина
оптической плотности атомного пара (А) в соответствии с основным законом
светопоглощения пропорциональна концентрации поглощающих частиц ( Сат ) -
атомов определяемого элемента в атомизаторе:
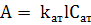 (1.6)
(1.6)
где 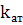 -
коэффициент поглощения света свободными атомами;
-
коэффициент поглощения света свободными атомами; 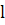 -
длина оптического пути (толщина слоя атомного пара).
-
длина оптического пути (толщина слоя атомного пара).
При постоянных условиях атомизации и заданном
режиме работы прибора концентрация атомов в атомизаторе  прямо
пропорциональна концентрации определяемого элемента в пробе (с). Таким образом,
можно записать:
прямо
пропорциональна концентрации определяемого элемента в пробе (с). Таким образом,
можно записать:
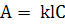 (1.7)
(1.7)
где k - коэффициент, включающий в себя как
собственно коэффициент поглощения ,
так и коэффициент перехода от к 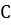 .
Как и в АЭС, коэффициент пропорциональности k является сугубо эмпирической
величиной, которая зависит от условий анализа и находится опытным путем
(градуировка).
.
Как и в АЭС, коэффициент пропорциональности k является сугубо эмпирической
величиной, которая зависит от условий анализа и находится опытным путем
(градуировка).
1.3.2. Атомизаторы
В ААС роль атомизатора состоит только в переводе
пробы в атомарное состояние, но не в возбуждении атомов. Поэтому рабочий
диапазон температур в ААС (около 800 - 3000 °С) в целом существенно ниже, чем в
АЭС. Основные типы источников атомизации - это пламенные и электротермические
(непламенные) атомизаторы.
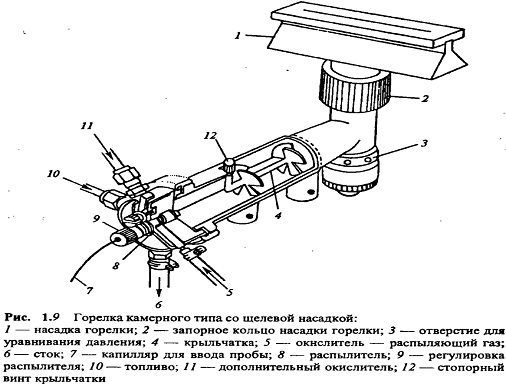
Пламя. Пламенный атомизатор для ААС представляет
собой горелку. Однако конструкции атомизаторов в АЭС и ААС различаются. В ААС
используют различные варианты щелевых горелок (рис. 1.9), в которых пламя имеет
форму вытянутой узкой щели. Этим обеспечивается большая длина оптического пути
и, в соответствии с уравнением (1.7), увеличение аналитического сигнала.
В ААС наиболее распространены следующие составы
горючих смесей: светильный газ - воздух (1500 - 1800 °С); ацетилен - воздух
(220 - 2300 °С); ацетилен - закись азота (2700 - 2950 °С).
Важнейшее достоинство пламенных атомизаторов -
высокая стабильность режима работы. Основной недостаток - низкая эффективность
атомизации, связанная с тем, что проба подается в атомизатор в виде раствора с
большой скоростью и, таким образом, находится в условиях высокой температуры
малое время.
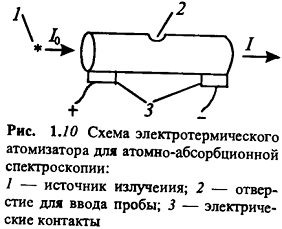
Электротермические атомизаторы. Способ
электротермической атомизации (ЭТА) в ААС изобретен Б.В. Львовым (1959 - 1960)
и в дальнейшем неоднократно совершенствовался. В настоящее время наиболее
распространенной конструкцией электротермических атомизаторов является
небольшая трубка (длина несколько сантиметров, внутренний диаметр до 1 см),
обычно графитовая, нагреваемая электрическим током большой силы (рис. 1.10). В
верхней части трубки имеется небольшое отверстие для ввода пробы. Жидкие пробы
вводят микрошприцем, возможен анализ и твердых проб. Для предотвращения
быстрого выгорания графита атомизатор помещают в атмосферу инертного газа -
обычно аргона высокой чистоты.
Электротермическая атомизация имеет много
преимуществ перед пламенной. Главное из них - значительное повышение
чувствительности определения вследствие увеличения эффективности атомизации.
Во-первых, проба находится в атомизаторе продолжительное время, а, во-вторых,
графит облегчает диссоциацию устойчивых оксидов многих элементов своими
восстановительными свойствами. Кроме того, резко сокращается объем пробы,
необходимый для анализа и чувствительность дополнительно повышается. Также
возможно вести измерения в вакуумной УФ-области (ниже 186 нм), в которой
находятся интенсивные линии поглощения ряда неметаллов (фосфор, мышьяк). В ЭТА
можно непрерывно изменять температуру атомизатора в пределах 20 - 2700 °С,
меняя силу тока нагрева.
1.3.3. Источники
излучения
В ААС новым моментом является наличие в приборе
источников внешнего излучения. Главное требование - высокая степень
монохроматичности излучения, обусловленная узкополосной структурой атомных
спектров поглощения (ширина линий порядка 10-3 - 10-2 им). В настоящее время в
ААС в качестве источников излучения наиболее распространены разрядные лампы с
полым катодом и безэлектродные разрядные лампы. Они являются источниками
линейчатых спектров.
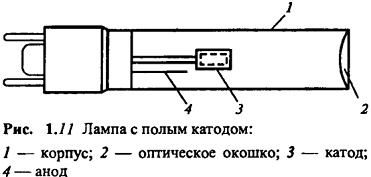
Лампа с полым катодом (рис. 1.11) представляет
собой стеклянный или кварцевый баллон, заполненный инертным газом под низким
давлением, внутри которого находятся два электрода - катод и анод. Катод имеет
форму чаши и изготовляется из чистого металла. При подаче напряжения на
электроды возникает тлеющий разряд с образованием положительных ионов
газа-наполнителя. Последние бомбардируют катод, выбивая атомы металла в газовую
фазу. Там эти атомы возбуждаются и испускают излучение, характерное для свободных
атомов соответствующего элемента. Таким образом, спектр излучения - это атомный
спектр материала катода (плюс линии возбужденных ионов газа-наполнителя). Из
него с помощью дифракционного монохроматора выделяют одну линию и используют ее
для атомно-абсорбционного определения соответствующего элемента.
Безэлектродные разрядные лампы также
представляют газоразрядные источники излучения. В такой лампе содержится
небольшое количество чистого вещества (или его летучего легкодиссоциирующегo
соединения), которое переводится в атомный пар и возбуждается под действием
микроволнового поля. Безэлектродные разрядные лампы изготовляют главным образом
для определения неметаллов (As, Se, Те, Р) и летучих металлов (Hg, Гb, Cs).
Серьезный недостаток разрядных ламп - их «узкая
специализация»: каждая лампа пригодна для определения только одного элемента.
Существуют, правда, и многоэлементные лампы, в которых катод изготовлен из
смеси (сплава) нескольких элементов, но у них эксплуатационные характеристики
хуже. Поэтому предпринимаются попытки создания источников излучения для ААС с
перестраиваемой частотой. Примеры таких источников - особо мощные (ксеноновые)
лампы, дающие непрерывный спектр, в сочетании с монохроматорами с высокой
разрешающей способностью, а также лазеры с перестраиваемой частотой: на
красителях и, на полупроводниковых диодах. Излучение последних отличается
высокой монохроматичностью, что позволяет определять даже изотопы элементов.
Тем не менее лампы с полым катодом и безэлектродные разрядные лампы до сих пор
используются в ААС наиболее широко.
1.3.4. Спектральные
помехи
Излучение фона. В ААС можно эффективно
скомпенсировать фоновое излучение атомизатора, используя модуляцию светового
потока источника. С помощью модулятора поток излучения направляют на пробу
периодическими импульсами. На рис. 1.12 показана временная диаграмма светового
потока на выходе из атомизатора. В момент времени t1
когда источник освещает пробу, регистрируется суммарная интенсивность (I
+ I0). В момент
времени t2 регистрируется
только фоновое излучение I0
Исправленное значение I
находится по разности.
Поглощение фона. Молекулы и микроскопические
частицы (фон) имеют широкополосные спектры поглощения и накладываются на узкие
линии поглощения атомов. Резкое различие в их ширине используют для компенсации
фонового поглощения. Для этого атомизатор и источник линейчатого спектра
одновременно освещают источником непрерывного спектра обычно дейтериевой
лампой, способ - дейтериевой коррекции фона. Излучение дейтериевой лампы
практически не поглощается свободными атомами, однако поглощается фоном.
Электронная система прибора разделяет сигналы от обоих источников света и
автоматически делает поправку на измеренное поглощение фона. Этим способом
удается скомпенсировать поглощение фона до 0,2 единиц оптической плотности.
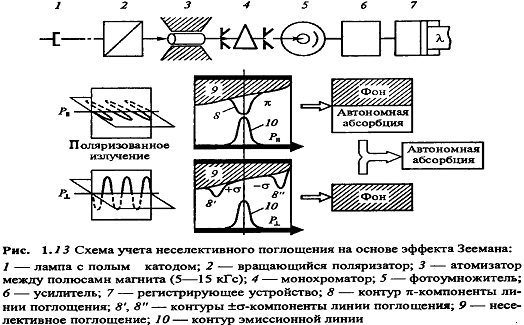
Более современный способ коррекции фонового
поглощения основан на эффекте Зеемана. Источник излучения или атомизатор
помещают между полюсами сильного электромагнита. В постоянном магнитном поле
атомная линия поглощения вследствие эффекта Зеемана расщепляется на три
компоненты. Положение одной из них, называемой π-компонентой,
совпадает с положением линии в отсутствие поля, а две другие (σ-компоненты)
симметрично смещены относительно нее в стороны больших и меньших частот (рис.
1.13). При этом наблюдается также поляризация света: π-компонента
поляризована в направлении, параллельно направлению магнитного поля, а σ-компоненты
- перпендикулярно. Полосы же фонового поглощения не расщепляются и не
поляризуются.
При использовании зеемановской коррекции
оптическую плотность измеряют при наложении магнитного поля в поляризованном
свете, применяя вращающийся поляризационный светофильтр. Когда направление
плоскости поляризации светофильтра совпадает с направлением магнитного поля, регистрируется
суммарное атомное (π-компонента)
неселективное поглощение. При перпендикулярном регистрируется только
неселективное поглощение. Чистое значение оптической плотности атомного пара
находят по разности. Зеемановская коррекция позволяет скомпенсировать
поглощение фона до 1-2 единиц оптической плотности.
Такие спектральные помехи, как наложение атомных
спектральных линий в ААС практически невозможны потому, что атомные спектры
поглощения беднее линиями, чем спектры испускания.
.3.5. Физико-химические
помехи
Физико-химические помехи в ААС имеют ту же
природу, что и в АЭС. Основными мешающими эффектами являются неполнота
атомизации и ионизация. Сходны и способы борьбы с ними - регулирование
температурного режима атомизации и применение спектроскопических буферов
(модификаторов матрицы). В ААС очень эффективным способом борьбы служит
программирование температуры атомизатора.
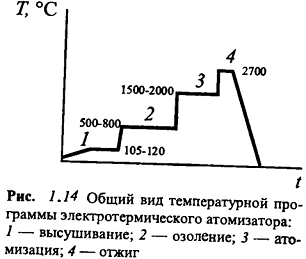
На рис. 1.14 приведен типичный вид такой
программы. Она состоит минимум из четырех стадий. В процессе высушивания
медленно испаряется растворитель. На стадии озоления из пробы удаляются все
компоненты более летучие, чем определяемый элемент. На стадии атомизации
измеряют аналитический сигнал. Заключительный кратковременный отжиг при
максимально возможной температуре необходим для регенерации поверхности
атомизатора, очистки его от труднолетучих компонентов.
1.3.6. Метрологические
характеристики и аналитические возможности метода
Чувствительность. Пределы обнаружения в ААС для
большинства элементов составляют 10-6 - 10-4 в пламенном и 10-9 - 10-7 % масс.
в электротермическом вариантах. ААС в целом чувствительнее, чем АЭС. В ААС
аналитический сигнал формируют атомы, находящиеся в основном состоянии, а в АЭС
- атомы в возбужденном состоянии, доля их весьма мала.
Диапазон определяемых содержаний в ААС
лимитируется величиной аналитического сигнала. Диапазон значений составляет от
нескольких сотых до 0,6 - 1,2 единиц оптической плотности. Проблемы с
определением малых значений А связаны со способом измерения оптической
плотности - по разности между интенсивностями падающего и прошедшего излучений.
При малых оптических плотностях эта разность
мала и погрешность велика. В областях высоких оптических плотностей погрешности
связаны с отклонениями от основного закона светопоглощения, вызванными
недостаточной монохроматичностью излучения источника, влиянием рассеянного
света и неоднородностью поглощающей среды. Малый диапазон определяемых
содержаний является существенным недостатком метода ААС.
Воспроизводимость в ААС (особенно в пламенном
варианте) несколько выше, чем в АЭС. Величины Sг
составляют для пламенного 0,005-0,05 и 0,02-0,1 для электротермического
способов атомизации. Улучшение воспроизводимости для ААС связано с тем, что
флуктуации температуры атомизатора почти не изменяют долю невозбужденных атомов
(она всегда близка к 100%), однако сильно влияют на долю возбужденных атомов.
Селективность в ААС часто бывает выше, чем в
АЭС. Это объясняется тем, что в ААС практически никакой роли не играет
наложение спектральных линий. Селективность в ААС лимитируется
физико-химическими помехами.
Главный недостаток метода ААС - трудность
осуществления многоэлементного анализа, поскольку для каждого элемента нужен
свой источник излучения. По этой же причине метод ААС непригоден для
качественного анализа.
Количественный анализ. Метод ААС - наиболее
чувствительный и удобный метод массовых одноэлементных определений большинства
металлов. Для количественного анализа методом ААС применяют методы внешних
стандартов (гpадуировочного графика) и добавок. Метод внутреннего стандарта
неприменим, т.к. ААС - одноэлементный метод анализа, не позволяющий
одновременно измерять аналитические сигналы двух элементов. Особенно широко в
ААС используют метод добавок. Потому, что помехи в ААС имеют физико-химическую
природу, т. е. являются с метрологической точки зрения мультипликативными.
Кроме того, ААС - это главным образом метод
анализа растворов. Для растворов, в отличие от твердых проб, метод добавок
легко реализуем технически.
1.4. Другие методы
атомной спектроскопии
.4.1. Атомно-флуоресцентная
спектроскопия
Метод атомно-флуоресцентной спектроскопии (АФС)
относится к числу люминесцентных. Аналитическим сигналом служит интенсивность
излучения, принадлежащего к оптическому диапазону и испускаемого возбужденными
атомами. Атомы возбуждаются под действием внешнего источника излучения. Доля
возбужденных атомов и, следовательно, интенсивность люминесценции I
определяются в первую очередь интенсивностью этого источника I0
в соответствии с приближенным соотношением
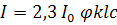 (1.8)
(1.8)
Как правило, квантовые выходы сильно уменьшаются
с ростом температуры. Ввиду того, что атомно-флуоресцентный анализ требует
высокой температуры, для свободных атомов величины 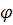 крайне
невелики. Поэтому в АФС решающее значение имеет использование как можно более
мощных источников излучения. В качестве таковых применяют высокоинтенсивные
разрядные лампы (с полым катодом или безэлектродные), а также лазеры с
перестраиваемой частотой.
крайне
невелики. Поэтому в АФС решающее значение имеет использование как можно более
мощных источников излучения. В качестве таковых применяют высокоинтенсивные
разрядные лампы (с полым катодом или безэлектродные), а также лазеры с
перестраиваемой частотой.
Сейчас метод АФС развивается в основном в
лазерном варианте (лазерная атомно-флуоресцентная спектроскопия, ЛАФС).
Использование лазеров позволило резко увеличить
чувствительность метода. Главное достоинство метода АФС - высокая селективность
(наивысшая среди методов оптической атомной спектроскопии), обусловленная
простотой спектров атомной флуоресценции и отсутствием наложения спектральных
линий различных элементов.
1.4.2. Рентгеновская
спектроскопия
Взаимодействие рентгеновского излучении с веществом.
При прохождении рентгеновского излучения через образец оно ослабляется
вследствие поглощения, а также упругого и неупругого (комптоновского) рассеяния
на электронах атомов твердого тела. Основной вклад в ослабление рентгеновского
излучения вносит его поглощение. С ростом длины волны (уменьшением энергии)
рентгеновского кванта массовый коэффициент поглощения постепенно увеличивается.
При достижении определенной длины волны края поглощения массовый коэффициент
ослабления резко уменьшается. Такой процесс повторяется многократно во всем
диапазоне длин волн (вплоть до вакуумного ультрафиолета).
Рентгеновский спектр - распределение
интенсивности рентгеновского излучения, испущенного образцом (РЭА, РФА) или
прошедшего через образец (РАА), по энергиям (или длинам волн). Рентгеновский
спектр содержит небольшое число спектральных линий (эмиссионный спектр) или
«скачков» поглощения (абсорбционный спектр). Фоновый сигнал эмиссионного
спектра формируют кванты рентгеновского излучения, неупруго рассеянные на электронах
атомов твердого тела. Рентгеновская эмиссия возникает при электронных переходах
между внутренними уровнями атомов. Относительная «простота» рентгеновского
спектра обусловлена ограниченным числом возможных электронных переходов.
Источники возбуждения спектра. Для возбуждения
спектра в РЭА, РАА и РФА используют рентгеновскую трубку.
Ее рабочим элементом является пара
вакуумированныx электродов - термоэмиссионный катод и охлаждаемый анод,
выполненный из тугоплавкого материала с хорошей теплопроводностью (W, Мо, Cu и
др). Анализируемый образец помещают непосредственно на анод рентгеновской
трубки. В результате бомбардировки электронами происходит эмиссия
рентгеновского излучения с поверхности образца. Для возбуждения спектра в РАА и
РФА используют первичное рентгеновское излучение, генерируемое рентгеновской
трубкой. В РАА степень монохроматичности рентгеновского излучения должна быть
выше.
Разновидностью РЭА является электроннозондовый
рентгеноспектральный микроанализ (РСМА). В нем для возбуждения рентгеновского
спектра используют моноэнергетический пучок электронов (анализ в «точке») или
сканирующий электронный пучок - растр (анализ участка поверхности). Таким
образом, РСМА - метод локального анализа. Источник возбуждения - электронная
пушка. Она состоит из авто- или термоэмиссионного катода и системы ускоряющих и
фокусирующих электростатических или магнитных линз, работающих в условиях
высокого вакуума.
Рентгеноэмиссионный анализ.
Аппаратурное оформление метода. Основными узлами
любого эмиссионного рентгеновского спектрометра (РЭА, РФА) являются источник
возбуждения спектра, входная щель (или коллиматор), устройство крепления и
ввода образца, выходная щель, обобщенная система анализа и детектирования
рентгеновской эмиссии. В зависимости от принципа работы последнего узла
различают спектрометры с волновой дисперсией (СВД) и спектрометры с
энергетической дисперсией (СЭД). В СВД для диспергирования рентгеновских лучей
используют кристалл-анализатор, а для их детектирования - пропорциональный или
сцинтилляционный детектор. В СЭД функции анализатора и детектора совмещает
охлаждаемый полупроводниковый детектор (ППД) К его достоинствам относятся
большая величина и меньшая длительность сигнала. СВД обладает более высоким
спектральным разрешением. Это позволяет уверенно различать в спектре линии с
близкими значениями длин волн. Однако СЭД обладает более высокой светосилой.
Это приводит к повышению интенсивности измеряемых спектральных линий.
Возможности метода и его применение. Метод РЭА
позволяет проводить одновременный многоэлементный качественный и количественный
анализ твердых образцов. С помощью СЭД можно определять элементы от Na до U, а
с помощью СВД - от В до U. Самые низкие величины определяемых содержаний
достигаются в случае тяжелых элементов в легких матрицах. Метод РСМА используют
для локального анализа поверхностных слоев образцов, содержащих
микроскопические гетерофазы (в том числе для анализа материалов высоких
технологий).
Рентгенофлуорисцентный анализ
Аппаратурное оформление метода. Схема
РФ-спектрометра и РЭ-спектрометра аналогичны. Вакуумные РФ-спектрометры могут
работать с длинноволновым рентгеновским излучением и определять легкие
элементы. Для локального анализа поверхности твердого тела применяют
современные РФ-спектрометры на основе капиллярной рентгеновской оптики.
Подготовка проб. Точность количественного РФА
определяется правильностью и надежностью подготовки проб. Могут быть
исследованы растворы, порошки, металлы и сплавы. Основное требование к пробе -
чтобы интенсивность аналитической линии определяемого элемента зависела от его
концентрации. Влияние всех остальных факторов должно быть исключено или
стабилизировано.
Возможности метода и его применение. Метод РФА
позволяет проводить неразрушающий одновременный многоэлементный качественный и
количественный анализ твердых и жидких образцов. Самые низкие величины
определяемых содержаний достигаются в случае тяжелых элементов в легких
матрицах. Метод РФА используют для анализа металлов, сплавов, горных пород,
экологического мониторинга почв, донных отложений.
Рентгеноабсорбционный анализ.
Аппаратурное оформление метода. Основными узлами
РА-спектрометра являются источник рентгеновского излучения, монохроматор,
Устройство крепления и ввода образца, детектор.
Возможности метода и его применение. Метод РАА
не нашел широкого применения из-за невысокой избирательности, но в тех случаях,
когда в матрице из легких элементов содержится только один определяемый элемент
большой атомной массы, применение данного метода вполне целесообразно. РАА
используется при серийных определениях тяжелых элементов в образцах постоянного
состава, например свинца в бензине и т.д.
1.4.3. Электронная
спектрометрия
Воздействие пучка рентгеновского излучения
(РФЭС) или электронов (ОЭС) приводит к эмиссии электронов с поверхности образца.
Электронный спектр представляет собой распределение эмитируемых электронов по
кинетическим энергиям. Эмиссия электронов происходит с внутренних электронных
уровней атома. Фоновый сигнал электронного спектра формируется неупруго
рассеянными электронами.
Важной характеристикой методов электронной
спектроскопии является глубина отбора аналитической информации. Поскольку
глубина проникновения рентгеновского излучения в РФЭС и электронного пучка в
ОЭС много больше глубины выхода эмитированных электронов, эффективная глубина
отбора аналитической информации определяется последним фактором. Критерием в
данном случае служит средняя длина свободного пробега (СДСП) фото- или
оже-электрона.
Рентгеновская фотоэлектронная спектроскопия.
Источник возбуждения спектра. Для возбуждения
электронного спектра в РФЭС используют характеристическое рентгеновское
излучение. Классический его источник - рентгеновская трубка. Для решения
специальных задач в качестве источника возбуждения спектра применяют
синхротронное излучение (СИ), возникающее при движении электрона с
релятивистскими скоростями и угловым ускорением в сильном магнитном поле.
Интенсивность СИ на 3-4 порядка выше интенсивности излучения рентгеновской
трубки. Перед использованием СИ монохроматизируют, в результате чего ширина
линии возбуждения составляет всего 0,2-0,3 эВ.
Аппаратурное оформление метода. Основные узлы
электронного спектрометра - источник возбуждения, устройство крепления и ввода
образца, энергоанализатор, детектор электронов - помещены в камеру, в которой
поддерживают сверхвысокий вакуум. В качестве детектора в электронной
спектроскопии используют электронный умножитель - пропорциональный счетчик
электронов. Его важной рабочей характеристикой является аппаратная функция.
Интенсивность электронной линии в спектре растет с увеличением значения
аппаратной функции.
Возможности метода и его применение. Метод РФЭС
позволяет проводить неразрушающий качественный и количественный элементный и
фазовый анализ поверхности твердого тела. Современные РФЭ-спектрометры комплектуют
также рентгеновскими трубками для локального анализа. Определять можно любые
элементы от Li до U. По
положению линий в электронном спектре можно однозначно идентифицировать
элементы (качественный анализ), а по интенсивности линий - определять их
содержание (количественный анализ). Методом РФЭС можно анализировать
поверхности разных материалов: металлов, сплавов, керамики, полимеров и др.
Метод РФЭС используют для решения проблем микроэлектроники, гетерогенного
катализа и т. д. (в том числе для анализа материалов высоких технологий).
Оже-электронная спектрометрия.
Источник возбуждения спектра. Для возбуждения
оже-электронногo спектра используют сфокусированный моноэнергетический
электронный пучок. Источником возбуждения служит электронная пушка с перестраиваемой
в широком диапазоне энергией электронов. Для построения карт распределения
элементов по поверхности образца применяют оже-электронкую микроскопию. Для
изучения тонкой структуры оже-электронных линий в качестве источника
возбуждения используют ренгеновское излучение - ОЭС с рентгеновским
возбуждением.
Аппаратурное оформление метода. Схема
оже-электронного спектрометра аналогична схеме рентгено-электронного
спектрометра. Однако необходимо дополнительно экранировать камеру анализа
спектрометра от внешних электромагнитных полей для защиты образца от внешних
вибраций. Ширина оже-линий составляет десятки эВ. К универсальным современным
энергоанализаторам относятся анализатор типа «цилиндрического зеркала» с
двойной фокусировкой и сферический энергоанализатор, обладающие высоким
пропусканием и разрешением.
Возможности метода и его применение. Метод ОЭС
позволяет проводить качественный и количественный элементиый и фазовый анализ
поверхности твердого тела. ОЭС - метод локального анализа. Определять можно
любые элементы от Li до U. По положению линий в электронном спектре можно
однозначно идентифицировать элементы (качественный анализ), а по интенсивности
линий определять их содержание (количественный анализ). «Тонкая структура»
оже-линии чувствительна к химическому окружению атомов и структуре поверхности.
РФЭС и ОЭС, относящиеся к методам анализа
поверхности твердого тела, занимают особое место в современной аналитической
химии. РФЭС и ОЭС - это классические методы анализа поверхности твердого тела.
С каждым годом возрастает число новых методов, расширяющих возможности РФЭС и
АЭС. Анализ поверхности и поверхностных слоев твердого тела - интенсивно
развивающееся направление современной аналитической химии.
Глава 2. Потенциометрия
В основе потенциометрических измерений лежит
зависимость равновесного потенциала электрода от активности (концентрации)
определяемого иона. Для измерений необходимо подобрать подходящий индикаторный
электрод и электрод сравнения, а также иметь прибор для измерения потенциала индикаторного
электрода в условиях, близких к термодинамическим, т. е. без отвода заметного
тока при замыкании цепи. Различают прямую и косвенную потенциометрию, или
потенциометрическое титрование.
.1. Индикаторные
электроды
.1.1. Мембранные
электроды
Возникновение потенциала мембранного электрода
обусловлено ионообменными процессами на границе раздела электрод-раствор,
потенциал металлического электрода определяется электронообменными процессами
на межфазной границе. По определению ИЮПАК, «ионоселективные электроды - это
сенсоры (чувствительные элементы, датчики), потенциалы которых линейно зависят
от 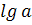 определяемого
иона в растворе».
определяемого
иона в растворе».
Важнейшей составной частью электродов является
полупроницаемая мембрана - тонкая пленка, отделяющая внутреннюю часть электрода
(внутренний раствор) от анализируемого и обладающая способностью пропускать
ионы только одного вида (катионы или анионы). Способность мембраны быть
проницаемой для ионов определенного знака заряда обусловлена наличием ионогенных
групп. Если мембрана контактирует с двумя растворами иона А+ с активностями a1
(анализируемый раствор) и a2
(внутренний раствор), то на внешней и внутренней сторонах мембраны происходит
обмен ионами.
Из-за различия активностей ионов А+ в растворе и
мембране на обеих сторонах мембраны возникают граничные потенциалы E1
и E2.
С помощью электродов сравнения, помешенных во
внешний и внутренний растворы, можно измерить разность E1
и E2 или так
называемый мембранный потенциал 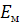 .
.
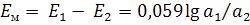
Активность ионов А+ во внутреннем растворе
постоянна, поэтому
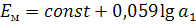
потенциал мембранного электрода линейно зависит
от логарифма активности иона А+ в анализируемом растворе.
Любая мембрана в той или иной мере проницаема
для всех ионов одного вида, находящихся в растворе, и поэтому необходимо
учитывать влияние посторонних ионов, например В+, на потенциал электрода. Ионы
В+ проникают в фазу мембраны в результате реакции обмена
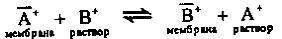
Константа равновесия этой реакции (константа
обмена, 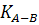 )
зависит от природы мембраны и природы иона В+. Подвижности ионов А и В, и
)
зависит от природы мембраны и природы иона В+. Подвижности ионов А и В, и 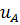 и
и
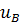 в
фазе мембраны различны, поэтому возникает диффузионный потенциал, вносящий
определенный вклад в величину .
в
фазе мембраны различны, поэтому возникает диффузионный потенциал, вносящий
определенный вклад в величину .
Потенциал мембранного электрода в растворе,
содержащем кроме определяемого иона А посторонние ионы В, С и другие,
описывается модифицированным уравнением Нернста (уравнением Никольского)
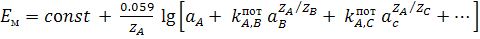
где 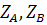 и
и
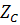 -
целое число, по знаку и величине равное заряду иона А, В и С (зарядовое число);
-
целое число, по знаку и величине равное заряду иона А, В и С (зарядовое число);
 -
потенциометрический коэффициент селективности;
-
потенциометрический коэффициент селективности;  -
включает значения потенциалов внешнего и внутреннего электродов сравнения и
зависит от природы мембраны, поскольку включает величину граничного потенциала
на внутренней стороне мембраны.
-
включает значения потенциалов внешнего и внутреннего электродов сравнения и
зависит от природы мембраны, поскольку включает величину граничного потенциала
на внутренней стороне мембраны.
Потенциометрический коэффициент селективности, 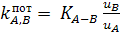 отражает
относительное влияние ионов А и В на величину мембранного потенциала и
характеризует способность мембраны различать ионы А и В, А и С и т.д. Основными
характеристиками ионоселективного электрода являются электродная функция,
селективность и время отклика. Электрод имеет нернстовскую электродную функцию
в интервале активности (концентрации), где зависимость потенциала от рА (-
отражает
относительное влияние ионов А и В на величину мембранного потенциала и
характеризует способность мембраны различать ионы А и В, А и С и т.д. Основными
характеристиками ионоселективного электрода являются электродная функция,
селективность и время отклика. Электрод имеет нернстовскую электродную функцию
в интервале активности (концентрации), где зависимость потенциала от рА (- )
линейна и имеет угловой коэффициент 59,16/
)
линейна и имеет угловой коэффициент 59,16/ мв
(25 °С). Протяженность этого интервала зависит от природы мембраны. При низких
концентрациях (для хороших электродов порядка 10-6 М) электрод утрачивает
электродную функцию (рис. 2.1); точка перегиба на графике характеризует
практическую величину предела обнаружения.
мв
(25 °С). Протяженность этого интервала зависит от природы мембраны. При низких
концентрациях (для хороших электродов порядка 10-6 М) электрод утрачивает
электродную функцию (рис. 2.1); точка перегиба на графике характеризует
практическую величину предела обнаружения.

Селективность электрода определяется величиной 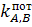 .
Если
.
Если 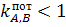 ,
электрод селективен относительно ионов А. Чем меньше числовая величина тем выше
селективность. Существуют различные способы оценки величины .
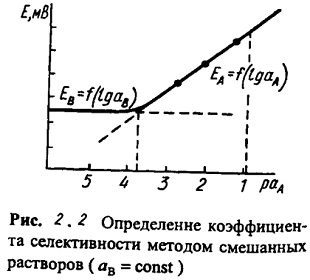
Чаще других. используют метод смешанных растворов, основанный на измерении
потенциала электрода в растворах с постоянной концентрацией мешающего иона В и
переменной концентрацией определяемого иона А. Точка пересечения линейных
участков полученной зависимости (рис. 2.2) дает величину
,
электрод селективен относительно ионов А. Чем меньше числовая величина тем выше
селективность. Существуют различные способы оценки величины .
Чаще других. используют метод смешанных растворов, основанный на измерении
потенциала электрода в растворах с постоянной концентрацией мешающего иона В и
переменной концентрацией определяемого иона А. Точка пересечения линейных
участков полученной зависимости (рис. 2.2) дает величину 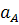 ,
по которой рассчитывают
,
по которой рассчитывают  .
.

Иногда используют метод отдельных растворов или
биионных потенциалов (рис. 2.3). Он основан на измерении потенциала электрода в
растворах, содержащих только ион А и только ион В. По экспериментальным данным
(кривые а и б соответственно) находят коэффициент селективности двумя
способами.
) Находят активности и
 ,
при которых электрод приобретает одинаковый потенциал в растворах А и В. В этом
случае
,
при которых электрод приобретает одинаковый потенциал в растворах А и В. В этом
случае 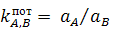 .
.
) Находят потенциалы E1
и E2, приобретаемые
электродом в растворах ионов А и В с одинаковой активностью по формуле

вычисляют коэффнциент селективности.
Следует заметить, что метод смешанных растворов
дает более надежные результаты и его использование предпочтительно.
Время отклика (время установления стационарного
потенциала) определяют по зависимости потенциала электрода от времени с момента
погружения в анализируемый раствор. В зависимости от природы мембраны время
отклика может колебаться от нескольких секунд до нескольких минут, зависит от
методики работы и изменяется от того, переносят ли электрод из более
концентрированного раствора в более разбавленный или наоборот. Чем меньше время
отклика, тем лучше, особенно при непрерывных измерениях в потоке или при
автоматизированных измерениях.
Ионоселективные электроды. Согласно рекомендациям
ИЮПАК, различают: первичные ионоселективные электроды - электроды с
кристаллическими мембранами, электроды с жесткой матрицей (стеклянные);
электроды с подвижными носителями - положительно заряженными, отрицательно
заряженными, незаряженными (с «нейтральными переносчиками»);
сенсибилизированные (активированные) электроды газочувствительные и ферментные.
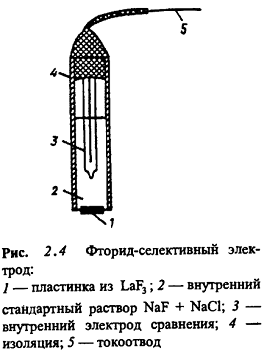
Электроды с кристаллическими мембранами.
Кристаллические гомогенные мембраны изготовляют из индивидуального кристаллического
соединения (LaF3 , Ag2S) или гомогенной смеси кристаллических веществ
(Ag2S+AgCl , Ag2S+CuS). При изготовлении гетерогенных кристаллических мембран
электродно-активное вещество смешивают с инертной матрицей (силиконовая смола)
или наносят на гидрофобизованный графит. Электрическая проводимость этих
мембран обусловлена способностью иона решетки с наименьшим радиусом и зарядом
перемещаться по вакансиям решетки. Для кристаллических мембран характерна
высокая специфичность, т.к. размер, форма и распределение заряда вакансии
решетки позволяют занять это место только определенному подвижному иону. Низкая
растворимость материала мембраны позволяет достигать очень низких пределов
обнаружения.
Превосходным электродно-активным кристаллическим
веществом является сульфид серебра, обладающий малой растворимостью (KS
~ 10-51), высокой устойчивостью к окислителям и восстановителям, низким
электрическим сопротивлением. Но наиболее совершенным электродом является
F-селективный электрод (рис. 2.4). Мембрана его выполнена из пластинки
монокристалла фторида лантана, активированного для увеличения дефектов решетки
(понижения электрического сопротивления) фторидом двухзарядного катиона (Ba,
Eu).
В настоящее время электроды с кристаллическими
мембранами делают и без внутреннего раствора, используя прямой контакт
металлического проводника и мембраны. Такие электроды называют твердотельными
(или электродами с твердым контактом), они удобнее в работе, чем электроды с
внутренним раствором.

Электроды с жесткой матрицей. Стеклянные
мембраны изготовляют из специальных стекол, подбирая их состав так, чтобы
мембрана проявляла повышенную селективность к определенному иону и позволяла
определять его в присутствии других. Первым ионоселективным электродом был
стеклянный электрод для измерения рН (рис. 2.5). В зависимости от целевого
назначения электрод может иметь разную форму и размер (от крошечных стерженьков
для введения в полость зуба или даже в отдельную клетку до шарика диаметром
1-15 мм для лабораторных аналитических работ).
В любом случае главной частью электрода является
тонкая рН-чувствительная мембрана. Обычно ее изготовляют из стекла, содержащего
22% оксида натрия, 6% оксида кальция и 72% оксида кремния. Внутренним раствором
служит 0,1 М раствор соляной кислоты, насыщенный хлоридом серебра.
Чувствительностью к ионам водорода обладает только хорошо вымоченная мембрана.
Специальными измерениями с изотопами доказано, что ионы 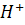 через
слой сухого стекла не проходят. Потенциал хорошо вымоченного стеклянного
электрода описывается уравнением
через
слой сухого стекла не проходят. Потенциал хорошо вымоченного стеклянного
электрода описывается уравнением
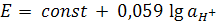 (2.2)
(2.2)
Таблица 3. Буферные смеси, рекомендуемые для
градуировки стеклянного электрода
|
Состав
буферной смеси
|
pH (25 °С)
|
|
Гидротартрат
калия (насыщ.)
|
3,557
|
|
KHC4H4O6 (0.05 M)
|
3,776
|
|
Гидрофталат
калия (0,05 М)
|
4,004
|
|
KH2PO4 + NaHPO4 (0,025 M)
|
6,863
|
|
Na2B4O7 (0,01 M)
|
9,183
|
|
NaHCO3 + Na2CO3 (0,025 M)
|
10,014
|
Стеклянный электрод пригоден для правильного
измерения рН в ограниченном интервале, зависящем от состава стекла. Изменяя
его, можно получать мембраны, обладающие высокой селективностью к другим ионам.
Созданы электроды для определения ионов натрия, калия и др.

Электроды на основе мембран с подвижными
носителями имеют жидкие мембраны - раствор ионообменника или «нейтрального
переносчика» в органическом растворителе, удерживаемый на пористом полимере
(рис. 10.16). Органический растворитель влияет на свойства электрода.
Современные конструкции подобных электродов
выполняют на основе пластифицированных мембран. Для их изготовления
электродно-активное вещество смешивают в определённых пропорциях с летучим
органическим растворителем. Из полученной пленки вырезают диск нужного диаметра
и приклеивают к тефлоновому корпусу.
Одним из лучших электродов такого типа является
К-селективный электрод с мембраной на основе «нейтрального переносчика»
валиномицина, пригодный для определения калия в присутствии большого количества
натрия. Его высокая селективность обусловлена соответствием размера внутренней
полости циклической молекулы валиномицина диаметру иона калия. Поэтому калий
прочно удерживается в полости.

Сенсибилизированные (активированные) электроды.
Газочувствительные электроды - это датчики, объединяющие индикаторный электрод
и электрод сравнения и имеющие газопроницаемую мембрану или воздушный зазор для
отделения анализируемого раствора от тонкой пленки промежуточного раствора
электролита. Он взаимодействует с определяемым газом, при этом изменяется
какой-то параметр промежуточного раствора, например рН, что и фиксирует
ионоселективный электрод. Отклик ионоселективного электрода пропорционален
парциальному давлению определяемого компонента в анализируемом газе.
Схематическое изображение гaзoчувствительногo электрода дано на рис. 2.7, в
табл. 4 приведены примеры практического применения.
Таблица 4. Применение газочувстительных
электродов
|
Определяемый
газ
|
Индикаторный
электрод
|
pH исследуемого
раствора
|
Состав
внутреннего раствора
|
Предел
обнаружения, М
|
|
CO2
|
Стеклянный
pH-чувствительный
|
<
4
|
10-2 M NaHCO3 + 10-2 M NaCl
|
10-5
|
|
NH3
|
То
же
|
< 12
|
10-2 M NH4Cl + 10-1 M KNO3
|
10-6
|
|
SO2
|
»
|
<
0,7
|
10-3 M NaHSO3 pH 5
|
5*10-6
|
|
HF
|
F-селективный
|
<2
|
1 M H+
|
10-8
|
|
H2S
|
S2--селективный
|
<5
|
Цитратный
буферный раствор, pH 5
|
10-7
|
Ферментные электроды - это датчики, в которых
ионоселективный электрод покрыт пленкой, содержащей фермент, способный вызвать
реакцию органического или неорганического вещества (субстрата) с образованием
веществ (ионов, молекул), на которые реагирует электрод. В основе работы
электрода лежит ферментативная реакция в результате, которой образуется
частица, обусловливающая отклик электрода.


Поэтому за изменением ее концентрации можно
проследить с помощью ионоселективного электрода. Селективность ферментных
электродов очень высока, поскольку каждый фермент катализирует только какую-то
определенную реакцию. На рис. 2.8 показана схема электрода для определения
мочевины, а в табл. 5 приведены другие примеры использования ферментных
электродов.
Таблица 5. Применение ферментных электродов
|
Субстрат
|
Фермент
|
Электрохимически
активная частица
|
Индикаторный
электрод
|
|
Пенициллин
|
Пенициллинназа
|
H+
|
Стеклянный
рН-чувствительный
|
|
Мочевина
|
Уреаза
|
NH4+ NH3
|
Стеклянный
NH4+-чувствительный
NH3-газовый
|
Алкогольоксидаза
|
CO2
|
CO2-газовый
|
|
Глюкоза
|
Глюкозооксидаза
|
H+
|
Стеклянный
рН-чувствительный
|
|
Фосфат-ион
|
Щелочная
фосфотаза
|
O2
|
Платиновый
|
2.1.2. Металлические
электроды
Различают активные и инертные металлические
электроды. Активные металлические электроды изготовляют из металлов, образующих
восстановленную форму обратимой окислительно-восстановительной системы (Ag, РЬ,
Cu, Cd). Потенциал такого электрода является функцией активности собственных
ионов в растворе, например для серебряного индикаторного электрода ( Ag+ + е ⇄
Ag)
Инертные металлические электроды изготовляют из
благородных металлов (Pt, Au).
Они служат переносчиками электронов от восстановленной формы к окисленной.
Поскольку их потенциалы зависят от соотношения активностей окисленной и
восстановленной форм полуреакции, их используют для индикации конечной точки
окиcлительно-восстановительногo титрования.
2.2. Измерение
потенциала
Как правило, ячейки с ионоселективными
электродами имеют очень высокое сопротивление, порядка 108 Ом. Для измерения
потенциала в таких случаях необходим электронный вольтметр с входным
сопротивлением на несколько порядков выше сопротивления ячейки. В противном
случае от ячейки будет отводиться заметный ток и измеренное значение потенциала
нельзя будет приравнять к значению равновесного потенциала.
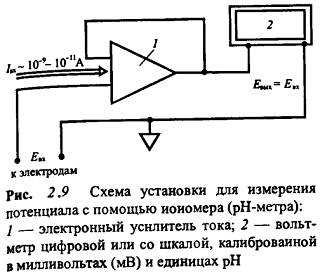
В аналитических лабораториях используют серийно
выпускаемые цифровые вольтметры или вольтметры со шкалой, калиброванной в мВ и
ед. рН. Эти приборы, называемые ионометрами или pH-метрами, имеют входное
сопротивление 1011 - 1012 Ом (рис. 2.9). Для более точных измерений потенциала
до сих пор применяют компенсационную схему, предложенную Погендорфом (1841).
Потенциометр Погeндорфа (рис. 2.10) состоит из
двух контуров верхнего, включающего источник постоянного напряжения, линейное
сопротивление АВ и переменное сопротивление R, и нижнего, состоящего из
линейного сопротивления АВ со шкалой, калиброванной в вольтах, скользящего
контакта С, двойного двухполюсного ключа П для введения в цепь стандартной
ячейки ЕCT или
изучаемой ЕX, нуль-инструмента
и телегpафного ключа К. В верхнем контуре постоянно течет ток.
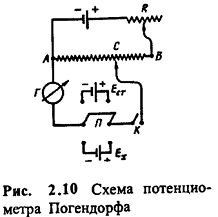
Перед измерением потенциала ЕX
настраивают потенциометр по элементу Вестона (ЕCT).
для этого ключом П в цепь вводят ЕCT,
устанавливают скользящий контакт на отметку 1,0183 В и подбирают сопротивление
R так, чтобы при кратковременном замыкании ключа К нуль-инструмент показал
отсутствие тока в нижем контуре. Затем ключом П вводят в цепь ЕX
и, замыкая ключ К на короткое время, перемещают скользящий контакт до тех пор,
пока в нижнем контуре не перестанет протекать ток. Поскольку шкала линейного
сопротивления градуирована в вольтах, остается записать искомую величину ЕX
.
2.3. Ионометрия
Раздел прямой потенциометрии, где индикаторным
электродом служит ионоселективный электрод, называют ионометрией. Это удобный,
простой и экспрессный современный метод: продолжительность анализа определяется
временем подготовки пробы, поскольку на само измерение тратится не более 1-2
мин. От других физико-химических методов ионометрия отличается простотой
методик и дешевизной измерительных приборов. Современные портативные ионометры
позволяют определять разнообразные ионы и растворенные газы не только в лаборатории,
но и в полевых условиях.
Уравнение для метода прямой потенциометрии,
связывающее величину измеренного потенциала электрода Е и рА. можно получить из
выражений

и 
где 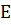 -
измеренный потенциал;
-
измеренный потенциал;  - потенциал
электрода сравнения;
- потенциал
электрода сравнения; 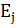 - диффузионный
потенциал;
- диффузионный
потенциал; 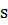 - yгловой
коэффициент (крyгизна) электродной реакции, 0,059/
- yгловой
коэффициент (крyгизна) электродной реакции, 0,059/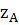 (25 °С). Решая систему уравнений, получаем
(25 °С). Решая систему уравнений, получаем
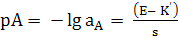 (2.3)
(2.3)
Здесь  включает
const, и
неизвестную величину поэтому нужно либо
оценить, либо исключить .
включает
const, и
неизвестную величину поэтому нужно либо
оценить, либо исключить .

Существуют три практических приема: метод
градуировки электрода, метод гpадуировочного графика и метод добавок. Самый
быстрый и простой из них - метод градуировки электрода. Чтобы оценить ,
достаточно измерить потенциал электрода в растворе с известным рА недостатки
этого метода: необходимость принимать найденную по уравнению (2.3) активность
равной концентрации (коэффициент активности неизвестен) и полагать неизменность
во
всех дальнейших измерениях, что весьма оптимистично. При построении
градyuровочного графика во все стандартные и анализируемые растворы вводят
одинаковый избыток индифферентногo электролита. Можно полагать, что ионная сила
всех растворов одинакова, и считать, что

Оптимальным, особенно в случае анализа растворов
сложного состава, является метод добавок, основанный на измерении потенциала
электрода в анализируемом растворе ( )
И после введения известного объема стандартного раствора ( Е 2 ). Так как
)
И после введения известного объема стандартного раствора ( Е 2 ). Так как
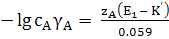
и 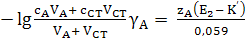
то 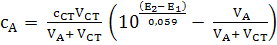
2.4. Потенциометрическое
титрование
Зависимость равновесного потенциала
индикаторного электрода от состава раствора, описываемую уравнением Нернста,
можно использовать для нахождения конечной точки титрования. Для этого измеряют
потенциал после добавления каждой порции титранта.
Заметив объем, при котором наблюдается резкое
изменение потенциала (скачок титрования), проводят точное титрование, для чего
прибавляют сразу почти весь необходимый объем титранта (на 1,5-2 мл меньше), а
затем добавляют его маленькими порциями (по 0,10 мл из микробюретки или по 2-4
капли из обычной бюретки) до достижения резкого изменения потенциала и еще
некоторый избыток. Из экспериментальных данных, записанных в виде таблицы,
можно методом численной интерполяции (найдя величины 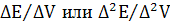 )
найти объем титранта, затраченный на достижение конечной точки. По полученным
данным можно построить кривую титрования в интегральной форме, в виде первой и
второй производных (рис. 2.11) и найти конечную точку гpафически. Во всех этих
случаях полагают, что кривая титрования симметрична относительно точки
эквивалентности, поскольку за конечную точку принимают точку максимального
наклона кривой. Если скачок титрования большой, то погрешность при невыполнении
этого допущения невелика.
)
найти объем титранта, затраченный на достижение конечной точки. По полученным
данным можно построить кривую титрования в интегральной форме, в виде первой и
второй производных (рис. 2.11) и найти конечную точку гpафически. Во всех этих
случаях полагают, что кривая титрования симметрична относительно точки
эквивалентности, поскольку за конечную точку принимают точку максимального
наклона кривой. Если скачок титрования большой, то погрешность при невыполнении
этого допущения невелика.
Гран предложил способ обработки данных
титрования без использования точки максимального наклона. Экспериментальные
данные преобразуют в функции, дающие линейную зависимость от объема титранта.
Этот способ позволяет получить более точные результаты для тех случаев, когда скорость
изменения р-функции вблизи точки эквивалентности мала и кривая титрования
выражена плохо.
Известен другой прием - титрование до заданного
потенциала. Если есть возможность оценить потенциал в конечной точке титрования
(это просто сделать, записав предварительно кривую титрования с помощью
автотитратора), можно воспользоваться и этим способом.
В потенциометрическом титровании применимы
кислотно-основные, окислительно-восстановительные реакции и реакции
комплексообразования, а также процессы осаждения, протекающие быстро и
количественно. Для кислотно-основного титрования в качестве индикаторного
применим любой электрод с водородной функцией: водородный, хингидрониый,
стеклянный.
Наиболее часто применяемый стеклянный электрод
подробно описан в предыдущем разделе.
Водородный электрод - это первичный электрод для
измерения рН. Потенциал водородного электрода зависит от активности ионов
водорода и измеряет в интервале рН от 0 до 14 (25 ОС) Сурьмяный электрод
пригоден для измерения рН в интервале от 3 до 10. Хингидронный электрод состоит
из платиновой пластинки, погруженной в насыщенный хингидроном (молекулярный
комплекс 1:1 хннона и гидрохннона) раствор. Электрод нельзя при менять при рН
> 9, а также в присутствии окислителей и восстановителей, реагирующих с
хиноном и гидрохиноном. Достоинством электрода является низкая погрешность
результатов.
Индикаторным электродом в
окислительно-восстановительном титровании служит платиновый электрод. Величина
скачка определяется разностью формальных потенциалов полуреакций. Желательно,
чтобы хотя бы одна из полуреакций была обратимой. При титровании не
рекомендуется измерять потенциал до прибавления титранта и вблизи точки
эквивалентности, так как в эти моменты из-за отсутствия одной из форм
(окисленной или восстановленной) полуреакции образуется смешанная
окислительно-восстановительная пара, где роль отсутствующей окисленной формы
выполняет растворенный кислород, а роль отсутствующей восстановленной формы -
вода. Приобретаемый электродом смешанный потенциал неустойчив, поэтому его
трудно измерить.
В осадительном титроваиии по реакции
галогенид-ионов с ионами серебра в качестве индикаторного пригоден серебряный
электрод. До точки эквивалентности потенциал электрода зависит от ахтивности
галогенид-ионов. За точкой эквивалентности при избытке ионов серебра потенциал
электрода определяется полуреакцией и зависит от активности собственных ионов.
Величина скачка зависит от растворимости осадка. Можно провести
дифференцированное титрование смеси хлорид-, бромид- и иодид-ионов.
Метод потенциометрического титрования имеет ряд
преимуществ перед прямой потенциометрией и титриметрией с визуальными
индикаторами. В отличие от прямой потенциометрии здесь не существует искажения
результатов за счет диффузионного потенциала, его влияние проявляется лишь в
смещенной кривой титрования вдоль оси потенциалов. Кроме того, нет
необходимости знать коэффициент активности определяемого иона.
К числу преимуществ перед визуальным титрованием
прежде вceгo относятся исключение субъективных ошибок, возможность анализа
мутных и окрашенных растворов, документальность и сравнительно легкая
автоматизация. Но, пожалуй, основное преимущество заключается в возможности
дифференцированного титрования компонентов смеси. Сочетание преимущества
инструментального фиксирования конечной точки и влияния органического
растворителя на кислотно-основные свойства позволяет, например, зафиксировать
раздельные скачки титрования для смеси пяти кислот - хлорной, соляной,
салициловой, уксусной и фенола, что совершенно невозможно сделать с помощью
индикаторов.
СПИСОК ЛИТЕРАТУРЫ
1. Золотов
Ю.А. Основы аналитической химии. Том 2. / Ю.А. Золотов, Е.Н. Дорохова, В.И.
Фадеева и др./Под ред. Ю.А. Золотов - М.: Высшая школа - 2004 - 503 стр.