|
соединение
|
бензол
|
пиррол
|
фуран
|
тиофен
|
|
Ароматичность %
|
100
|
37
|
12
|
45
|
|
Энергия
резонанса (ER) кДж/моль
|
150
|
90
|
68
|
122
|
Исходя из представления о p-избыточности пиррола и его
электронных аналогов, логично предположить, что эти соединения особенно склонны
к участию в реакциях с электрофилами. Это и наблюдается в действительности.
Свойства соединения, содержащие в цикле пиррольный гетероатом, можно сравнить
со свойствами анилина, в молекуле которого аминогруппа тоже активирует
ароматическое ядро.
4.
Шестичленные гетероциклы - азины и их аналоги
Пиридин представляет собой электронный аналог бензола, в
котором одна группа СН (метиновая группа) заменена атомом азота. В отличие от
пиррола, атом азота в нейтральной молекуле пиридина образует две s - и одну
p-связь, т.е. вносит в ароматический секстет один электрон. Неподеленная пара
электронов атома азота в сопряжение вступать не может, потому что ось ее
орбитали ориентирована в пространстве перпендикулярно осям орбиталей p-электронов атомов углерода.
Этот тип атома называется "пиридиновый". Находясь в составе кольца
атом азота пиридинового типа не может быть донором, он является акцептором
p-электронов, так как азот более электроотрицателен, чем углерод. Это
иллюстрируют канонические структуры пиридина:
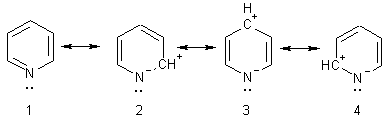
Индуктивный и мезомерный эффекты атома азота в пиридине действуют
в одном направлении (-I - и - M), смещая электронную плотность к атому азота. Это является
причиной того, что на атомах углерода индуцирован частичный положительный заряд
и электронная плотность в ядре понижена. Поэтому пиридин относят к типу
p-дефицитных ароматических гетероциклов. Наибольший положительный заряд
сосредоточен в a - и g-положениях. Здесь просматривается аналогия с электронным
строением нитробензола, имеющего частичные положительные заряды в орто -
и пара-положения.

Атомы кислорода и серы также могут быть атомами
"пиридинового" типа. Наличие такого атома в цикле обусловливает
существование катионных изоэлектронных аналогов бензола - солей пирилия и
тиопирилия. Положительно заряженные атомы кислорода и серы, как и пиридиновый
атом азота вносят в p-систему гетерокольца один электрон и обладают
неподеленными электронным парами, которые в сопряжении с p-электронной системой
кольца участия не принимают. Ввиду того, что электроакцепторные свойства атома
с полным положительным зарядом больше, чем нейтрального, соли пирилия и
тиопирилия значительно более p-дефицитны, нежели электронейтральный пиридин.

Шестичленные гетероциклы с несколькими гетероатомами также более
p-дефицитны, чем пиридин. Особенно заметно это ощущается тогда, когда атомы
азота расположены в b-положении друг к другу, например, в пиримидине и симм-триазине.
Причина заключается в том, что в этих случаях каждый гетероатом независимо от
другого наводит положительный заряд на одних и тех же атомах углерода, как в
случае согласованной ориентации, например, в мета-динитробензоле.
гетероциклическое органическое соединение пятичленное

Из вышеизложенного очевидно, что пиридин, ди - и триазины и,
особенно соли пирилия, должны легко вступать в реакции с нуклеофильными
реагентами и быть пассивными по отношению к электрофилам.
5. Азолы
В молекулах диазолов (пиразола и имидазола) имеются
гетероатомы как "пиррольного", так и "пиридинового" типов,
в связи с чем соединения такого типа в рамках концепции p-избыточных (пиррол) и
p-дефицитных (пиридин) гетероциклов называют p-амфотерными. Среди полярных
граничных структур, описывающих состояние молекул имидазола и пиразола, имеются
структуры как с положительными, так и с отрицательными зарядами на атомах
углерода.

В действительности, химическое поведение азолов иллюстрирует их
амфотерность - они способны к реакциям и с электрофилами, и с нуклеофилами.
6. Пиррол
Основность
Неподеленная пара электронов пиррольного азота в значительной
степени вовлечена в циклическое p-сопряжение, она малодоступна и поэтому пиррол
проявляет весьма низкую основность (pKa сопряженной кислоты = -
3,8). Расчеты показывают, что среди возможных катионов пирролия
термодинамически наиболее выгоден резонансно-стабилизированный катион I - результат
протонирования атома углерода в a-положениях. N-катион III наименее
стабилен, т.к. во-первых, заряд в нем сосредоточен на одном атоме, и,
во-вторых, нарушена ароматическая система сопряжения: это - фактически диен.
Катион II занимает промежуточное положение.

Тем не менее, в кислой среде возможно протонирование всех атомов
ядра. Кристаллические соли, соответствующие катионам типа I, могут быть выделены при пропускании
сухого HCl через растворы полиалкилпирролов в инертных растворителях.
Доказательством образования катиона III является легкий дейтерообмен протона при пиррольном атоме азота в
кислой среде. Несмотря на то, что катион III наименее устойчив, он образуется и разрушается быстрее, чем
катионы I и II, поэтому NH-протон
пиррола дейтерируется быстрее, чем CH-протоны.
Это явление называется кинетической основностью. Кинетическая основность азота
всегда выше, чем углерода. С-катион II отвечает за процесс полимеризации пиррола в кислой среде, когда
образуется полимер переменного строения "пиррол-красный". Механизм
первых стадий этой реакции подтверждается строением выделенного тримера.

Склонность пирролов к полимеризации под действием кислот накладывает
серьезные ограничения на участие пирролов в реакциях с электрофилами, т.к. эти
превращения протекают зачастую в кислой среде.
Реакции по атому азота
Кислотность пиррола (рКа 17,0) близка к кислотности
этанола (рКа 15,9) и сильные основания способны превратить его
пиррил-ион, который представляет собой высоко p-избыточный гетероаналог
циклопентадиенила. Получаемые действием амидов металлов или щелочных металлов
натриевые и калиевые соли пиррола легко взаимодействуют с электрофилами -
алкилируются и ацилируются по атому азота, тогда как смешанные N-пиррилмагнийгалогениды (связь N-Mg менее ионная, чем N-Na) реагируют
преимущественно по a-положению ядра.

Кинетический продукт ацилирования N-ацилпиррол в отсутствии катализатора при нагревании
перегруппировывается в более устойчивый термодинамический продукт -
2-ацетилпиррол.
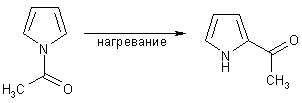
Реакции по атомам углерода
В нейтральной и кислой среде пирролы почти никогда не реагируют с
электрофилами по атому азота. Электрофильная атака направляется, главным
образом, в a-положения ядра. Это объясняется тем, что образующиеся при этом
s-комплексы типа I, как и в случае протонирования, наиболее
стабильны среди всех возможных.

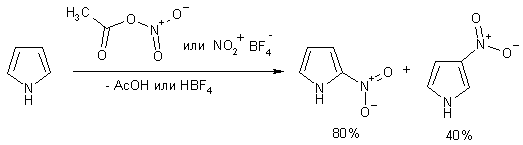
Нитрование
Нитрующая смесь вызывают быстрое разложение пиррола, поэтому для
нитрования используют специальные реагенты: ацетилнитрат, приготавливаемый
заранее из 70% -ной HNO3 и уксусного ангидрида, либо кристаллический тетрафторборат
нитрония в неводных растворителях. Во втором случае (более мягкий реагент)
выходы выше. Соотношение a - и b-изомеров составляет примерно 4: 1.
Сульфирование
Сульфирование пиррола по причине его ацидофобности олеумом
невозможно; однако, пиррол-2-сульфокислота образуется с хорошим выходом при
использовании комплекса SO3 с пиридином, который называется
пиридинсульфотриоксидом.

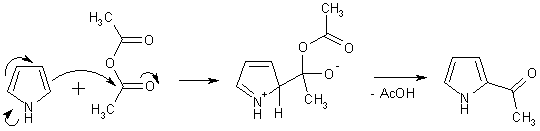
Ацилирование
Ацилирование пирролов по атомам углерода, в отличие от бензола,
не требует применения катализаторов, используемых обычно в реакции
Фриделя-Крафтса. Пиррол настолько активен, что реагирует при нагревании с
уксусным ангидридом, при этом легко могут быть получены как 2-ацил-, так и
2,5-диацилпирролы.
Алкилирование пиррола по Фриделю-Крафтсу редко применяется в
синтетических целях, т.к. при этом быстро образуются полиалкилпроизводные.
Галогенирование
Взаимодействие пирролов с молекулярными галогенами приводит, как
правило, к замещению всех атомов водорода при свободных С-атомах, в то же
время, сульфурилхлорид при охлаждении монохлорирует пиррол в a-положение.

Моногалогенпирролы, в отличие от полизамещенных соединений,
неустойчивы. Галогенирование пирролов протекает столь активно, что зачастую
сопровождается отщеплением заместителей, например, карбоксильной группы. В свою
очередь атом галогена, чаще всего иода, легко удаляется при гидрировании. Это
позволяет получить незамещенное положение в ядре в том случае, когда более
доступным в качестве исходного соединения оказывается замещенный пиррол,
например:

Пиррол, обладающий высокой нуклеофильностью легко вступает в реакции
с такими слабыми электрофилами, с которыми бензол не реагирует даже в жестких
условиях. Например, пиррол намного легче, чем даже фенолы, вступает в реакцию
карбоксилирования по Кольбе - достаточно нагревания с карбонатом аммония.

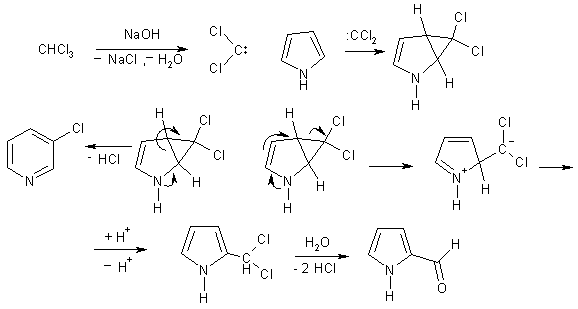
Пиррол, как и фенол, формилируется в условиях реакции
Реймера-Тимана, когда в качестве активного реагента выступает дихлоркарбен.
Однако данное взаимодействие осложняется параллельным процессом - происходит
расширение цикла в результате внедрения дихлоркарбена в одну из p-связей
пиррольного ядра, что приводит к 3-хлорпиридину. Объяснение этого состоит в
том, что промежуточно образуется производное циклопропана, которое
стабилизируется двумя альтернативными путями. Соотношение продуктов зависит от
условий проведения реакции.
В слабокислой среде пиррол сравнительно устойчив, что позволяет
ввести его, например, в реакцию азосочетания, которая еще раз подтверждает его
высокую p-избыточность. Если пиррол вводить во взаимодействие с солью диазония
в слабощелочной среде (реагирует пиррил-ион), то можно получить 2,5-бис
(фенилазо) пиррол.

Пиррол способен к конденсации с карбонильными соединениями своим
a-положением, причем результат реакции зависит от природы альдегида или кетона.
Если реакция с формальдегидом и алифатическими альдегидами в кислой среде дает,
в основном, полимеры, то при конденсации с ацетоном основным продуктом
оказывается метилированный порфириноген. Взаимное отталкивание в пространстве
метильных групп способствует планаризации интермедиата - тримера, поэтому в
ходе следующей стадии легче образуется циклический тетрамер, чем линейный.

При взаимодействии пирролов с ароматическими альдегидами по
аналогичному механизму образуются порфириногены, которые, однако,
самопроизвольно окисляются кислородом воздуха в ароматические мезо-тетраарилпорфирины.

В случае конденсации пиррола с пара-диметиламинобензальдегидом
в слабокислой среде может быть выделен первичный продукт конденсации -
красно-фиолетовый катион арилиденпирроления (цветная реакция Эрлиха).

Для получения незамещенных порфириногена и порфирина пиррол
целесообразно сначала превратить в свободное основание Манниха, а уже затем
проводить конденсацию. Порфириноген под действиекм большинства окислителей,
например, при нагревании в хлороформе с хлоранилом, превращается в незамещенный
порфирин - порфин.

7. Индол
Индол представляет собой конденсированную биядерную систему,
состоящую из ядра пиррола и бензола. Систематическое название индола - бензо [b] пиррол. Химические
свойства пиррола и индола во многом схожи, но имеются и различия.
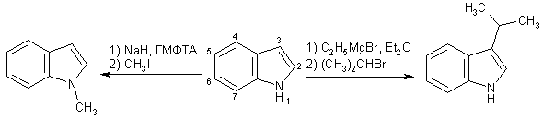
Как и пиррол, индол обладает NH-кислотностью (pKa " 17), его N-анион, генерируемый
сильными основаниями (EtONa, t-BuOK, и т.д.), проявляет схожую с пиррил-ионом активность: натриевые и
калиевые соли алкилируются и ацилируются по азоту, тогда как смешанные
магнийгалогенидные N-производные - по атому С (3), т.е. по b-положению, но не по
a-углеродному атому, как это происходит в пирроле.
Последнее обстоятельство объясняется тем, что в анионе, как и в
нейтральной молекуле индола, отрицательный заряд сосредоточен в большей степени
на углеродном атоме в положении 3, чем на С (2) - атоме. Это легко легко видеть
в наборе резонансных структур, описывающих N-анион индола:
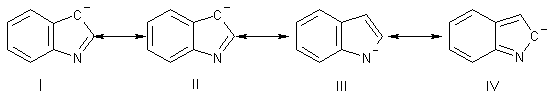
Очевидно, что перенос заряда на атом 2 невыгоден (структура IV), потому что при этом нарушается
ароматичность бензольного ядра, тогда как структуры I и II содержат ароматическое бензольное кольцо.
Наличие электроноакцепторных заместителей в положении 3 сильно
повышает кислотность, и алкилирование можно проводить в присутствии значительно
более слабых оснований.

Индол проявляет высокую активность в реакциях с разнообразными
электрофилами, причем замещение ориентируется также в положение 3, но не по
a-углеродному атому, как это имеет место в пирроле. Резонансные структуры для
s-комплексов индола с участием a - и b-углеродных атомов приводят к тем же
выводам, что вышеприведенная схема для N-анионов: s-комплекс, образующийся в результате присоединения
электрофила к атому 3, более выгоден.

Нитрование
Индолы, не имеющие заместителей в положениях 2 и 3, аналогично
пирролу полимеризуются под действием сильных кислот, поэтому нитрование таких
соединений проводят слабыми нитрующими реагентами - этилнитратом в присутствии
метилата натрия (реагирует анион индола) или бензоилнитратом в нейтральной
среде

-Метилиндол более устойчив в кислой среде, чем индол, поэтому он
успешно нитруется в жестких условиях действием азотной кислоты. В молекулу
можно ввести до трех нитрогрупп, однако для синтеза чистого
мононитропроизводного следует использовать ацилнитраты.

Интересно протекает реакция 2-метилиндола с нитрующей смесью:
ввиду того, что соединение нацело протонировано, реакция по гетероциклическому
ядру не идет вовсе, а промежуточный ковалентный аддукт с серной кислотой
реагирует в сопряженное с атомом азота положение 5 бензольного ядра.

Сульфирование
Сульфирование индола, как и пиррола, проводят действием не
кислотного реагента пиридинсульфотриоксида. Если в положении 3 присутствует
заместитель, например, метильная группа, то реакция ориентируется в положение
2.

Галогенирование
Галогенирование индолов протекает очень легко в положение 3,
однако получаемые галогениндолы не устойчивы к действию кислот, поэтому для
успешного галогенирования используют реагенты, в ходе реакции с которыми HHal по возможности не выделяется:
N-бромсукцинимид (NBS), SO2Cl2, KI3, пербромид пиридиния.
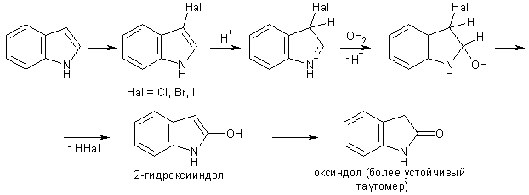
Если при С-атоме в положении 3 стоит заместитель, то первоначально
по этому атому образуется катионный аддукт с галогеном, который затем
трансформируется в результате нуклеофильной атаки растворителем, что приводит к
2-оксииндолам либо их производным.

Кроме перечисленных выше превращений, индолы способны вступать в
реакции электрофильного ацилирования, формилирования по Вильсмайеру,
азосочетания и конденсации с карбонильными соединениями. Все реакции идут в
мягких условиях и ориентируются в положение 3.
Примеры:

Замещающая группа в положении 3, как правило, не препятствует
электрофильной атаке в это положение и превращение завершается замещением этой
группы электрофилами (ипсо-замещение).

Исключение составляют соли диазония, которые в эту реакцию не
вступают.
8. Фуран
Из трех рассмотренных пятичленных гетероциклов фуран является
наименее ароматичным и во многих реакциях заметно проявляется его диеновый
характер.
Реакции электрофильного ароматического замещения для фурана
известны, однако они требуют специальных реагентов, т.к. под действием
протонных кислот фурановое кольцо разрушается намного легче, чем пиррол и тем
более индол.
А в общих чертах, фуран в этих реакциях значительно похож на
пиррол - весьма активен и реагирует преимущественно a-положениями.

Сульфирование
Сульфирование фурана, как и пиррола, можно провести с помощью
пиридинсульфотриоксида в органическом растворителе. В реакции образуется небольшая
примесь фуран-2,5-дисульфокислоты.

Выделение фуран-2-сульфокислоты осуществляют, разрушая
образующийся 2-фурилсульфонат пиридиния карбонатом бария, и получают
нерастворимую бариевую соль.
Нитрование
Как и пиррол, фуран нельзя подвергать действию нитрующей смеси,
однако можно использовать ацетилнитрат в пиридине. Реакция идет медленнее, чем
в случае пиррола, причем промежуточно образуется ковалентный продукт
присоединения реагента в положения 2 и 5.

Фураны, имеющие электроноакцепторные заместители, менее
ацидофобны, поэтому они нитруются и сульфируются обычными реагентами.

Галогенирование
Взаимодействие фурана с галогенами (бромом и, особенно, с хлором)
протекает бурно и приводит к образованию полигалогенпроизводных.
Моногалогенфураны могут быть получены лишь в мягких условиях, например,
действием диоксандибромида. Существуют разногласия по поводу механизма реакций
галогенирования фурана: не исключено, что они протекают как присоединение
молекулы галогена к положениям 2 и 5 и последующее отщепление молекулы
галогеноводорода.

В качестве подтверждения механизма присоединения-отщепления
рассматривается действие брома на фуран в метанольном растворе, в результате
которого образуется 2,5-диметокси-2,5-дигидрофуран.
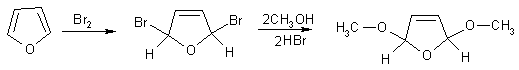
Промежуточное образование аддуктов доказывается также на примере
реакции бромирования 2,5-дибромфурана.

Формилирование
Формилирование фурана по Вильсмайеру протекает также гладко, как и
в случае пиррола, тогда как ацилирование требует обязательного прибавления
катализатора Фриделя-Крафтса.
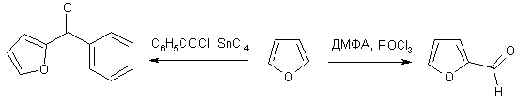
Реакция Дильса-Альдера
Существуют превращения, в которых проявляется диеновый характер
фурана. Наиболее характерным превращением является реакция диенового синтеза
(Дильс-Альдер). Сам фуран и многие его производные легко реагируют с малеиновым
ангидридом, дегидробензолом и другими диенофилами.
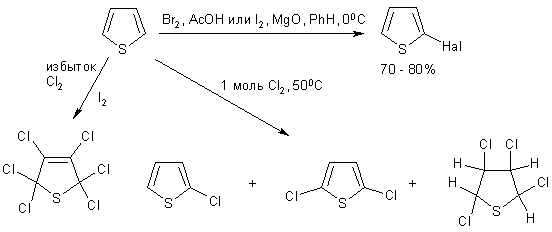
9. Тиофен
Среди рассматриваемых гетероциклов тиофен является наиболее
ароматичным и по своим свойствам во многом напоминает бензол. Производные
тиофена сопутствутствуют производным бензола в продуктах каменноугольной смолы
и весьма на них похожи, зачастую имеют даже похожий запах.
Тиофен намного устойчивее к действию кислот, чем пиррол и
фуран, поэтому он может быть введен в разнообразные реакции электрофильного
замещения. которые ориентируются в a-положение.

При нагревании со 100% -ной H3PO4 тиофен тримеризуется, реакция начинается с образования
a-протонированного катиона, который подвергается атаке нейтральной молекулой
тиофена.

Сульфирование
С электрофилами тиофен реагирует по механизму обычного
ароматического электрофильного замещения. Его можно сульфировать серной
кислотой при комнатной температуре, что применяется для выделения тиофена из
каменноугольного бензола в промышленности.

Нитрование
Для мононитрования тиофена в a-положение лучше всего применять
борфторид нитрония: ацетилнитрат дает до 20% примеси 3-нитротиофена, тогда как
азотная кислота реагирует слишком бурно, иногда со взрывом. После появления в
ядре одной нитрогруппы активность понижается настолько, что дальнейшее
нитрование требует применения дымящей HNO3.

Галогенирование
Тиофен легко бромируется и иодируется (в отличие от фурана) в
a-положение. Действие хлора приводит к смеси продуктов.
Ацилирование
Тиофен ацилируется по Фриделю-Крафтсу только в присутствии кислот
Льюиса, причем реакция сопровождается частичным осмолением из-за
самоконденсации образующихся кетонов.

Реакция Вильсмайера протекает с хорошим выходом, но при высокой
температуре. Промежуточную соль иминия можно выделить.

Конденсация с карбонильными соединениями
Аналогично пирролу и фурану, тиофен активно конденсируется с
карбонильными соединениями, однако реакцию редко удается остановить на стадии
первичного карбинола, зачастую получаются ди-, три - и полимеры.

В отличие от фурана, разлагающегося в этих условиях, тиофен может
быть с неплохим выходом превращен в бис-хлорметильное производное
действием большого избытка формалина и HCl.

Заслуживает внимания важная с исторической точки зрения цветная
"индофениновая реакция" тиофена с изатином в присутствии серной
кислоты.

Интенсивно-синий индофенин стал причиной открытия тиофена. До 1882
г. считалось, что индофениновая реакция свойствена ароматическим углеводородам,
т.к. использовавшийся тогда каменоугольный бензол всегда содержал примесь
тиофена. Однако однажды этот красивый опыт не удался на лекции В. Мейера, т.к.
в тот раз он использовал синтетический, а не каменноугольный, бензол. Стало
ясно, что цветную реакцию давала примесь, которая позднее была выделена и
идентифицирована как новое соединение - тиофен.
Расширение цикла
Для замещенных тиофенов известна реакция расширения цикла, не
характерная для пиррола и фурана; механизм этого превращения не известен.
Интересно, что в результате наблюдается довольно редкое явление - превращение
ароматического соединения в антиароматическое.

Реакции по атому серы
Тиофен вступает в своеобразные реакции по атому серы:
алкилирование и окисление надкислотами.

Если соли S-алкилтиофения
являются устойчивыми и, судя по спектральным и химическим свойствам,
ароматическими соединениями, то S,S-диоксиды тиофена не ароматичны, и могут быть получены
только для a,a-дизамещенных субстратов. Эти соединения реагируют с диенофилами.

10.
Взаимопревращение пятичленных гетероциклов
В жестких условиях пиррол, фуран и тиофен способны к
раскрытию кольца под действием нуклеофилов, поэтому при наличии подходящего
реагента они способны переходить друг в друга, что можно объединить на схеме:

Эта реакция носит имя Юрьева, она протекает при 350°С в
присутствии катализатора Al2O3.
11. Пиридин
Превращения по атому азота
Пиридин - основание средней силы с величиной рКа=
5,2, измеренной в воде, (основность алифатических аминов колеблется в интервале
pKa 9-11). Пиридин образует кристаллические соли с большинством
протонных кислот и часто применяется как основный катализатор или растворитель,
способствующий связыванию кислот, выделяющихся в ходе той или иной химической
реакции.

Как нуклеофил, пиридин реагирует с алкилгалогенидами и другими
алкилирующими реагентами по механизму SN2 или SN1 в зависимости от природы субстрата.

Соли N-алкилпиридиния являются ароматическими
соединениями, т.к. неподеленная электронная пара, использующаяся для
образования новой связи, не участвует в ароматическом сопряжении. Эти
устойчивые вещества проявляют склонность к реакциям с нуклеофилами из-за
высокой p-дефицитности. Например, нуклеофильное гидроксилирование действием
едкого кали дает a-пиридоны.

При взаимодействии пиридинов с ацилгалогенидами образуются N-ацилиевые соли. Эти соединения мало устойчивы, и легко
гидролизуются обратно.

Именно по причине низкой устойчивости ацилпиридиниевые соли имеют
важное значение для синтетической химии как мягкие ацилирующие реагенты.
Например, отметим О-ацилирование кетоенолов - реакция протекающая по необычному
механизму с промежуточным образованием аддукта по a-положению гетероцикла.

При обработке пиридина и его производных надкислотами образуются N-оксиды пиридинов, о свойствах которых будет сказано
отдельно.

Реакции по атомам углерода
Взаимодействие с электрофилами
Как было показано выше, пиридин представляет собой p-дефицитное
соединение, поэтому взаимодействие с электрофилами для него не характерно, тем
более, что электрофильные реакции протекают в кислой среде, где во
взаимодйствие с электрофилом вступае пиридиниевый ион, обладающий еще большей
p-дефицитностью. Эти реакции протекают значительно труднее, чем в бензоле.
Пиридин атакуется только сильнейшими электрофилами, причем в весьма жестких
условиях. Электрофильное замещение при ориентируются в положение 3, что
напоминает ориентацию SE2-реакций
в нитробензоле. Такая ориентация легко объясняется сравнительной устойчивостью
грначных структур, описывающих катионные s-комплексы возникающие в результате
присоединения электрофила к g-, b-, и a-положениям пиридинового кольца.
Очевидно, что только s-комплекс II не
содержит вклада структуры с положительно заряженным атомом азота.

Как уже сказано, сначала происходит атака электрофила или протона
по азоту, что дополнительно пассивирует субстрат, переводя его в катион. Если
предотвратить комплексообразование по гетероатому, то реакция атомов ядра с
электрофилами протекает более легко.
Нитрование
Нитрование собственно пиридина, особенно, нитрующей смесью, когда
гетероцикл нацело протонирован, протекает чрезвычайно трудно и практического
значения не имеет.

,6-Диметил - и 2,4,6-триметилпиридины, аминопиридины и пиридоны
намного активнее нитруются в форме катионов.

,6-Дигалогенпроизводные пиридина, которые как основания слабее
пиридина, реагируют легче, т.к. пиридиновый атом азота протонирован в меньшей
степени - концентрация свободного основания больше. Нитрование протекает легче
в случае использования апротонного нитрующего реагента, например:

Сульфирование
Сульфирование пиридина серной кислотой протекает несколько легче,
чем нитрование, но тоже возможно лишь в жестких условиях. При еще более высоких
температурах возможна перегруппировка в 4-сульфокислоту.

Любопытный продукт может быть получен при сульфировании 2,6-ди-трет-бутилпиридина.
Собственно сульфирование протекает легко, т.к. объемные заместители
препятствуют комплексообразованию SO3 по атому азота. Образующаяся 2,6-ди-трет-бутилпиридин-3-сульфокислота
при нагревании превращается в циклический сульфон за счет одной из метильных
групп трет-бутильного заместителя.

Галогенирование
Пиридины могут быть прогалогенированы. Ввести атом иода с
удовлетворительным выходом не удается, однако бром - и хлорпиримидины синтезируются
получены весьма просто.
Оксиды пиридина проявляют большую активность в реакциях с
электрофилами, чем сам пиридин. Существуют два типа электрофильного замещения в
N-оксидах: первый вариант (без добавления
нуклеофила) приводит, в основном, к N-оксидам
4-замещенных пиридинов. Это на первый взгляд странное обстоятельство связано с
замечательной электронной природой N-оксидной
функции, которая может проявлять себя одновременно не только как акцептор, но и
как донор электронов. Это иллюстрирует приведенная ниже схема.

Поэтому N-оксид любого
замещенного пиридина, имеющего свободным положение 4, можно нитровать дымящей HNO3 с хорошим выходом.

Другой подход к использованию N-оксидов - проведение реакции в присутствии слабых нуклеофилов,
которые могут входить в состав реагента, например, ацетилнитрата. При этом N-оксид превращается в неароматический аддукт, который и
атакуется электрофилом. Можно сказать, что пиридиновый атом азота на время
становится донором электронов. Реакция позволяет получать 3-нитро - и
3,5-динитропиридины с хорошими выходами.

Аналогично происходит вступление в положение 5 еще одной нитрогруппы
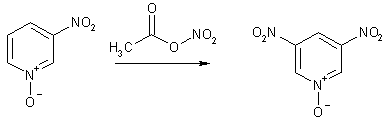
Нуклеофильное замещение
Характерными превращениями пиридина являются реакции
нуклеофильного замещения. Реакция аминирования пиридина при нагревании с амидом
натрия (реакция Чичибабина) приводит к образованию a-аминопиридина.
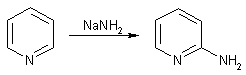
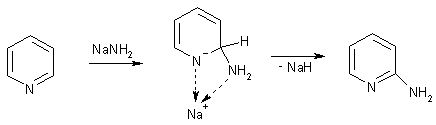
Реакция замещения атома водорода в пиридине аминогруппой действием
амида натрия всегда ориентируется в положение 2. Превращение имеет сложный
механизм: ходе реакции выделяется молекулярный водород, что заставляет
предположить промежуточное образование гидрида натрия. Этот факт, а также
отсутствие продуктов замещения водорода в положении 4 объясняют предварительной
координацией атома натрия с пиридиновым атомом азота.
Обработка пиридинов свежеплавленным едким кали приводит к
гидроксилированию до a-пиридонов, которые являются более устойчивой формой
существования a - и g-гидроксипиридинов.

Реакции нуклеофильного замещения галогена в пиридине протекают по
таким же двум альтернативным механизмам, как и в галогенаренах -
присоединение-отщепление (АЕ) и отщепление-присоединение (ЕА). Реакция АЕ
(присоединение нуклеофила с образованием s-комплекса, и отщепление уходящей
группы) протекают преимущественно по атомам 2, 4 и 6, в которых сосредоточен
максимальный положительный заряд. Кроме того, атом азота участвует в
делокализации отрицательного заряда соответствующих s-комплексов, как
нитрогруппа в случае нитрохлорбензолов. Легко видеть, что наиболее стабильными
являются анионные s-комплексы I и III.

С помощью реакции типа нуклеофильного АЕ в молекулу пиридина можно
ввести самые разнообразные заместители. Вот несколько примеров:

оксиды пиридина и N-алкилпиридиниевые соли вступают в те же
реакции легче самого пиридина. Особенно интересны N-оксиды a-галогенпиридинов, сразу превращающиеся при действии
некоторых нуклеофилов в конденсированные биядерные гетероциклы, образование
которых протекает с участием N-оксидной
группы.

Если галоген находится в положении 3, то реакция пиридинов с
нуклеофилами чаще идет по механизму ЕА через промежуточное образование
гетероаналога дегидробензола - гетарина.

Свободнорадикальные реакции
При действии атомарных хлора и (высокие температуры) брома на
пиридин происходит свободнорадикальное галогенирование, которое, в отличие от
электрофильного, ориентируется в положения 2 и 6.

Для препаративных целей имеют значение реакции пиридина с
нуклеофильными радикалами (реакция Миниши). Источниками радикалов служат
различные органические соединения в присутствии перекисей и соли железа (II), катион которого служит в качестве
переносчика электронов.
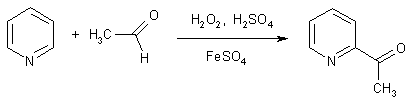
Механизм реакции включает стадии гомолитического разложения
перекиси, превращения реагента в свободный радикал и его присоединение к
пиридину и последующую ароматизацию.

Таким путем в положения 2 и 4 пиридина и хинолина можно ввести
гидроксиметильную группу, диалкиламидную и другие функциональные группы.
Литература
1.
Артеменко А.И., Тикунова И.В., Ануфриев Е.К. Практикум по органической химии. -
М.: Высшая школа, 2007-187с.
.
Березин Б.Д., Березин Д.Б. Курс современной органической химии. Учебное пособие
для вузов. - М.: Высшая школа, 2001. - 768с.
.
Глинка Н.Л. Общая химия/ Под ред.В.А. Рабиновича. - Л.: Химия, 1986. - 704с.
.
Градберг И.И. Практические работы и семинарские занятия по органической химии.
- М.: Дрофа, 2011. - 352с.
.
Сборник задач по органической химии. Учебное пособие/ Под ред.А.Е. Агрономова.
- М.: Изд-во МГУ, 2010. - 160с.