Влияние валентного состояния металла-добавки на термоокислительную стойкость ингибированного полиэтилена
МИНИСТЕРСТВО
ОБРАЗОВАНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
Учреждение
образования
«Гомельский
государственный университет
имени
Франциска Скорины»
Биологический
факультет
Кафедра
химии
Допущено к защите
Зав. кафедрой
__________ Дроздовой Н.И.
« _____ »
______________ 2013г
Влияние
валентного состояния металла-добавки на термоокислительную стойкость
ингибированного полиэтилена
Дипломная
работа
Исполнитель:
студентка группы БИ
- 52
В.С.Береснева
Научный
руководитель
Е.В.Воробьева
к.х.н., доцент
Гомель
2013
Содержание
Введение
Обзор
литературы
.1
Процессы окисления полиолефинов
.2
Влияние переходных металлов на окисление полиолефинов
.3
Краткая характеристика ингибиторов окисления
.4
Механизм действия антиоксидантов
Программа,
объект и методика исследования
.1
Программа исследований
.2
Характеристика основного объекта исследований
.3
Материалы и оборудование
.4
Методика проведения эксперимента
Результаты
исследований и их обсуждение
.1
Окисление полиэтилена, содержащего фенольный и аминный антиоксиданты
.2
Влияние валентности металла оксида на окисление полиэтилена, ингибированного
фенольным антиоксидантом
.2
Влияние валентности металла оксида на окисление полиэтилена, ингибированного
аминным антиоксидантом
Заключение
Список
использованных источников
Введение
Постоянно растущие требования к качеству изделий
из пластмасс предопределяют создание новых полимерных материалов, обладающих
стабильными свойствами и улучшенными эксплуатационными характеристиками. При
переработке, эксплуатации и хранении полимеры подвергаются воздействию ряда
химических и физических факторов, которые приводят к старению и потере
исходного комплекса физико-химических свойств. К основным физическим факторам
относятся тепло, свет, различные механические нагрузки и др. В зависимости от
конкретных условий эксплуатации эти факторы могут оказывать одновременное или
раздельное воздействие на полимер, инициируя при этом различные
физико-химические процессы, ухудшающие качество полимера вплоть до его
разрушения [1].
Так, практически все природные и синтетические
полимеры подвержены окислительному старению. В связи с этим разработка способов
стабилизации и создание путем модификации полимеров с комплексом
физико-химических свойств, стабильным в процессах переработки, хранения, эксплуатации
является одной из важнейших задач физики и химии полимеров.
В настоящее время используют два подхода к
решению проблемы повышения стабильности свойств полимерных материалов: синтез
новых высокомолекулярных соединений и направленная модификация промышленно
выпускаемых полимеров путем введения небольших количеств различных ингредиентов
(стабилизаторов-модификаторов). Второй подход является более перспективным как
с экономической, так и с технологической точек зрения [2].
Стабилизаторы, препятствующие развитию
окислительных реакций в полимерах, называют антиоксидантами. По механизму
действия антиоксиданты делятся на две большие группы. Первую группу составляют
вещества (ингибиторы), которые реагируют со свободными полимерными и радикалами
на стадии их образования. К этой группе относятся широко применяемые на
практике соединения, на основе ароматических аминов и фенолов с разветвленными
алкильными заместителями. Ко второй группе относятся вещества, способные
вызывать разложение образующихся первичных продуктов окисления -
гидропероксидов (к ним относятся сульфиды, меркаптаны, тиофосфаты и др.) [3].
Эффективную защиту от термоокислительного
старения обеспечивает применение пары антиоксидантов, действующий по разным
механизмам, взаимоусиленный стабилизирующий эффект смесью двух антиоксидантов
называют синергизмом.
По механизму стабилизации полимеров можно
выделить цепную и нецепную стабилизацию. Первая связана с дезактивацией
активных центров цепного процесса (цепное ингибирование), вторая - с
дезактивацией веществ, участвующих в любых реакциях в полимере, приводящих к
его старению (нецепное ингибирование). Примером последнего способа стабилизации
полимеров может служить образование металлами-катализаторами неактивных
комплексных соединений [4].
Стабилизация структуры полиолефинов, поиск для
этой цели новых антиоксидантов, повышение эффективности уже существующих всегда
были актуальными вопросами химии высокомолекулярных соединений. Особенно остро
проблемы стабилизации структуры полимера и эффективности антиоксидантов встают
при ингибировании полиолефинов, контактирующих с переходными металлами
(металлополимерные материалы и конструкции; вторичный полиолефин, загрязненный
переходными металлами в процессе эксплуатации и рециклинга). Известно, что
соединения переходных металлов являются катализаторами окислительных процессов
полимера, что приводит к существенному сокращению эксплуатационного периода
металлополимерной конструкции или изделия. По этой причине использование
антиоксидантов для ингибирования полиолефинов в составе металлополимерных
систем обычно сопровождается снижением эффективности добавки. Однако,
экспериментально установлено, что в некоторых металлополимерных системах
ингибирующая способность АО может «аномально» - неаддитивно изменяться: либо
повышаться, либо очень резко снижаться.
Поэтому целью дипломной работы является изучение
изменений эффективности фенольного и аминного антиоксидантов в условиях
окисления полиолефинов, контактирующих с переходными металлами разной
валентности.
Задачами являются:
изучение основных закономерностей процесса
окисления (старения) полимеров;
изучение влияние металлов (и их оксидов) на
процессы окисления ингибированного полиэтилена;
проведение эксперимента, выявляющего влияние
оксидов (Co2O3 и Co3O4; CrO3 и Cr2O3; FeO и Fe2O3; Sb2O3 и Sb2O5, СuO и Cu2O)
на изменения эффективности фенольного антиоксиданта ирганокса 1010;
проведение эксперимента, выявляющего влияние
оксидов (СuO и Cu2O) на изменения эффективности аминного антиоксиданта неозона
Д.
окисление полимер фенольный
антиоксидант
1. Обзор литературы
.1 Процессы окисления полиолефинов
Как и все органические материалы, полимеры
подвержены окислению. Это приводит к изменению вязкости, цвета, охрупчиванию
изделии ухудшению физико-механических характеристик. Окисление происходит на
каждой стадии существования полимерного материала - при его производстве и
хранении, при переработке в изделия и последующем использовании. Окисление
также называют деструкцией или старением. Обычно разделяют термомеханическую (в
процессе переработки) и термоокислительную (эксплуатация изделия) деструкцию
полимерного материала. Разные полимерные материалы обладают различной
стойкостью к старению - например, полипропилен сильно подвержен деструкции даже
при комнатной температуре, а полистирол и полиметилметакрилат (оргстекло)
стабильны даже при температурах переработки.
Процесс взаимодействия полимеров (и других
органических соединений) с кислородом называется автоокислением, процесс
автоокисления необратим и состоит из трёх стадий: инициирование; рост и
разветвление; передача и обрыв цепи. В большинстве случаев процесс окисления
полимеров характеризуется наличием индукционного периода, в течение которого не
происходит видимых изменений[5].
Окисление полимеров протекает по
свободнорадикальному механизму, начальная стадия которого в обобщенной форме
описывается следующей кинетической схемой.
Зарождение цепей:
(инициатор) → r• → P• (1.1)
PH + O2
→ P• + HO2•
(1.2)
2PH + O2 → P• + H2O2 + P•
(1.3)
Продолжение цепи:
P• + O2 → PO2• (1.4)+ PH →
POOH + P• (1.5)+ CHR=CHR'→ ROOCHRC=CHR' (P•) (1.6)
Вырожденное разветвление цепи:
POOH → PO• + HO• (1.7)+ PH →
PO• + H2O + P• (1.8)+ CHR=CHR' → PO• + HOCHRC∙HR' (P•) (1.9)
Обрыв цепей:
P• + P• → PP (1.10)• + PO2• →
POOP (1.11)• + PO2• → O2 + POOP (1.12)
Образование свободных радикалов и макрорадикалов
на стадии инициирования может происходить под влиянием содержащихся в полимере
примесей, остатков инициаторов полимеризации, а также при действии света,
механических напряжений, при нагревании и т. д. Дополнительное количество
свободных радикалов образуется в результате распада полимерных пероксидов и
гидропероксидов по реакциям (1.7)-(1.9). Развитие цепи происходит при
взаимодействии пероксидного радикала POO• с полимером (реакция 1.5).
Пероксидный радикал стабилизируется, отрывая подвижный атом водорода от
молекулы полимера; при этом вновь образуется макрорадикал, взаимодействующий с
кислородом.
Окисление имеет вырожденно разветвленный
характер, который состоит в том, что образующийся по реакции (1.4) гидроперокеид
POOH нестабилен и распадается с образованием новых свободных радикалов PO•,
НО•, P•, POO•, которые также отрывают атомы водорода от молекулы полимера.
В результате скорость присоединения кислорода и
окисления полимера резко возрастает; это явление называется автокатализом.
Кинетические кривые поглощения кислорода полимерами имеют S-образную форму,
характерную для всех автокаталитических (самоускоряющихся) процессов
представлены на рисунке 1.
[O2]
Рисунок 1 - Кинетические кривые поглощения
кислорода в процессе окисления нестабилизированного(1) и стабилизированного (2)
полимеров
Обрыв цепи происходит в результате реакций
рекомбинации или диспропорционирования. Рекомбинация макрорадикалов с концевыми
пероксидными группами происходит с высокой скоростью (в отличие от медленной
рекомбинации обычных макрорадикалов в твердом полимере). В аморфных полимерах
выше температуры стеклования и в кристаллических выше температуры плавления
скорость рекомбинации таких макрорадикалов резко возрастает.
Следует отметить, что в отличие от
низкомолекулярных углеводородов, при окислении которых все радикалы P•
превращаются в пероксидные POO•, в окисляющемся полимере часть алкильных
макрорадикалов вступает в реакции изомеризации, передачи цепи и др [6].
Скорость окислительной деструкции определяется
скоростью диффузии кислорода в полимер и скоростью химического взаимодействия
полимера с кислородом. Скорость диффузии кислорода наиболее высока, если
полимер находится в растворе или расплаве. При температурах ниже температуры
стеклования, а тем более кристаллизации, доступ кислорода в полимер затруднен.
Чем выше степень кристалличности полимера, тем меньше скорость диффузии
кислорода. Растяжение и ориентация полимера также замедляют диффузию кислорода.
Однако в этом случае окисление ускоряется за счет механической активации.
Низкой скоростью диффузии кислорода характеризуются сетчатые полимеры [7].
Механизм и скорость процесса окисления зависят
также от строения макромолекул. Так, ненасыщенные высокомолекулярные соединения
окисляются значительно быстрее, чем насыщенные, поскольку в них легче
образуются пероксиды, благодаря чему ускоряется стадия инициирования.
При окислении ненасыщенных полимеров кислород
может присоединяться по двойной связи с последующим разрывом макромолекулы или
образованием гидропероксидов [8]. Они обнаруживаются по ИК-спектрам пленок
окисленных полимеров: в спектрах поглощения появляются полосы, характерные для
свободных и связанных гидропероксидных групп[4].
В первом случае реакция протекает по схеме [8]
O2
~CH2-CH=CH-CH2~
~CH2-CH=CH-CH2~
~CH2-CH-CH=CH~
| |
OO∙ OOH
~CH2-CH-CH-CH2~
~CH2-CH-CH-CH2~
| | | |
O - O O· O∙
~CH2-CH +CH-CH2~ (1.13)
|| ||
O O
Возможно также непосредственное образование
гидропероксидов ненасыщенных углеводородов с присоединением кислорода к
метиленовой группе, находящейся в α-положении
по отношению к двойной связи:
O2
~CH2-CH=CH-CH2~
CH2-CH=CH-CH~
|
OOH (1.14)
Первичным продуктом окисления полипропилена,
содержащего третичный атом углерода, также является гидропероксид (1.15).
OOH
O2 |
~CH2-CH-CH-CH2~
~CH2-CH-CH-CH2~ (1.15)
| | |
|
CH3 CH3
CH3 CH3
Обрыв цепи при окислительной деструкции в
результате рекомбинации радикалов может приводить к образованию межмолекулярных
связей. Так, при окислительной деструкции полиэтилена получаются нерастворимые
сшитые структуры (1.16):
·
~CH2-CH2-CH-CH2-CH2~
~CH2-CH2-CH-CH2-CH2~
· |
~CH2-CH2-CH-CH2-CH2~ ~CH2-CH2-CH2-CH-CH2~
(1.16)
Следует отметить, что при окислительной
деструкции наблюдается изменение состава полимера в результате появления
карбонильных, карбоксильных и других кислородсодержащих групп.
Карбонильные группы образуются как при
фотохимическом, так и термическом окислении полимеров и легко обнаруживаются
методом ИК-спектроскопии. Их образование объясняется распадом алкоксирадикалов,
которые возникают в актах обрыва, продолжения и вырожденного разветвления
цепей, а также распадом гидропероксидных групп [9].
Состав карбонильных групп существенно
различается при термическом и фотохимическом окислении полиэтилена: при
термическом образуются преимущественно кетонные группы, в то время как при
фотохимическом образуется смесь кетонных, альдегидных, карбоксильных и
сложноэфирных групп. Образование этих групп при фотоокислении связано с
фотораспадом карбонильных групп.
Наряду с функциональными группами при окислении
полимеров образуются и низкомолекулярные продукты: СО, СO2, Н2O, углеводороды,
альдегиды, кислоты, ацетон [4].
Окисление полимера сопровождается сшивкой
макромолекул, изменением надмолекулярной структуры и деструкцией, что
отражается на физико-механических (эксплуатационных) свойствах образцов
полимеров. Поскольку полимеры получаются в промышленных условиях, они неизбежно
загрязняются примесями металлов в процессе получения. Примеси металлов попадают
в полимер или в виде продуктов превращения металлосодержащих катализаторов, или
из-за контакта полимера с металлической аппаратурой. Кроме того при
практическом использовании полимеров их наполняют металлами и их соединениями,
а это отражается на их окислительной стойкости [2].
Процессы окисления полимеров классифицируют по
характеру зависимости поглощения кислорода от продолжительности окисления:
линейные - поглощение кислорода начинается сразу
и происходит с относительно постоянной скоростью;
автотормозящие - кислород быстро поглощается в
начале реакции с постепенным замедлением в последующем;
автокаталитические - скорость поглощения
кислорода возрастает при протекании процесса;
комбинированные - можно рассматривать
одновременно как автокаталитические и автотормозящие.
Большинство полимеров окисляются по четвертому
типу с индукционным периодом и автоускорением.
Исходя из анализа научной литературы, можно
сказать, что каталитическая активность металлов переменной валентности связана
с их склонностью участвовать в окислительно-восстановительных реакциях и
разрушать гидропероксид с образованием свободных радикалов. Причем,
каталитическое действие соединений переходных металлов проявляется, только при
условии их участия в химических превращениях в форме ионов [10].
Выделяют несколько направлений каталитического
участия ионов металлов по следующим схемам:
Ускорение разложения гидропероксидов на
свободные радикалы в результате обмена электронами:
Me n+ + ROOH ®
Me (n+1)+ + OH- + RO• (1.17)(n+1) + ROOH ® Me n+ + H+
+ RO2• (1.18)
По мнению большинства исследователей, это
основная возможность проявления металлами каталитического влияния на процесс
окисления.
Активация кислорода, в результате которой
происходит, взаимодействие возникшего ион-радикала с гидропероксидом,
вызывающее ускорение разложения последнего:
Me n+ + O2 ®
Me (n+1)+ + O2-• (1.19)- •+ ROOH ® RO• + OH- +
O2 (1.20)
III. Взаимодействие иона металла и молекулярного
кислорода с образованием активного комплекса, который интенсивно
взаимодействует с углеродной цепью, генерируя свободный радикал:
O2 + Men+ « [Me ( n+1)+ + O -2 ]  aR• (1.21)
aR• (1.21)
Однако в
научной литературе приведен ряд фактов, указывающих на то, что контакт
полиолефинов с переходными металлами и их соединениями может приводить к
стабилизации структуры полиолефинов, т.е. к ингибированию процесса окисления
(например, медь и свинец). Такое положение объясняется следующим. Реакциям
разложения гидропероксидов обычно предшествует комплексообразование -POOH
входит во внутреннюю координационную сферу металла, а распад комплекса
протекает по двум путям - гомолитическому и гетеролитическому [4].
Преобладание
гетеролитического распада приводит к низкой эффективности инициирования. В
зависимости от того, какое направление распада преобладает, соединение металла
ускоряет или замедляет окисление.
Полимерные
материалы в процессе эксплуатации или хранения могут контактировать с
агрессивными средами, под действием которых протекают следующие процессы:
сорбция компонентов агрессивной среды; десорбция из полимерного материала различных
добавок (стабилизаторов, пластификаторов и т.д.); химическая деструкция;
растворение полимера; изменение физической структуры (степени кристалличности,
микропористости и т.д.). Под действием этих процессов происходит изменение
(чаще всего ухудшение) эксплуатационных свойств полимерных материалов:
механических, оптических, диэлектрических, сорбционных, диффузионных и т.п.
Окисление
твердых полиолефинов имеет много общего с окислением низкомолекулярных
углеводородов. Также как и в случае низкомолекулярных углеводородов, окисление
полиолефинов сопровождается образованием свободных радикалов. Но образованные
радикалы имеют особенность - возможность перемещения свободной валентности с
одного фрагмента макромолекулы на другой. Это явление получило название эстафетного
механизма миграции свободной валентности [11].
Эстафетный
механизм более эффективен, но для его реализации необходима достаточно высокая
химическая активность радикалов, что зависит от строения фрагмента, несущего
свободную валентность, строения полимера и температуры. Диффузия атомов и
низкомолекулярных радикалов может успешно конкурировать с эстафетным
механизмом, но для этого необходим достаточно интенсивный источник этих
радикалов. Когда такой источник в виде Н2 или С12 специально вводится в
полимер, то нетрудно получить доказательства реализации именно этого механизма
[12].
Эстафетный
механизм подтверждается многочисленными данными, в том числе и методом ЭПР. В
окисляющемся полимере непрерывно происходят превращения типа: РO2 •→Р•→РO2→Р•→
РО • →Р • и т. д., т. е. налицо эстафета химических превращений. В
промежутках между ними сегмент макромолекулы смещается за счет теплового
движения всех сегментов макромолекулы. Поэтому эстафетный механизм
представляется наиболее естественным для окисляющихся полимеров. С изменением
условий эстафетный механизм миграции свободной валентности может уступить место
механизму с участием низкомолекулярных радикалов. При температурах выше 330 К в
полиолефинах сегментальная подвижность настолько высока, а химическая реакция
идет настолько быстро, что в отсутствие активных низкомолекулярных добавок
реализуется преимущественно эстафетный механизм. При 300 К и ниже в
полиолефинах часто реализуется перенос свободной валентности с участием
низкомолекулярных радикалов. Условия эксперимента влияют на реализацию
конкретного механизма гибели радикалов [1].
Зависимость
скорости автоокисления от парциального давления кислорода отражает существенное
отличие окисления полимера в твердой фазе от окисления жидких углеводородов. Окисление
углеводородов не зависит от парциального давления кислорода, как в присутствии
инициатора, так и в режиме автоокисления, когда инициатором служит образующийся
гидропероксид [2]. Обусловлено это тем, что в жидкости алкильные радикалы (при
не очень малом парциальном давлении кислорода и интенсивном перемешивании)
очень быстро реагируют с кислородом, так что выход РООН в расчете на
поглощенный кислород близок к 100 %, а соотношение между одиночными и блочными
группами зависит только от конкуренции интра- и интермолекулярной реакций
продолжения цепей с участием РО2•. В полимере из-за медленной микродиффузии
кислорода реакция Р• с O2 протекает гораздо медленнее, так что даже в
кинетической области окисления, когда обрыв цепей происходит по реакции РO2• с
РO2•, проявляется конкуренция реакций.
• →
•ОРОН (1.22)
Р• + РН→
РН + Р• (1.23)
Это
отражается на составе гидропероксидных групп.
Выше уже
отмечалось, что распад гидропероксидных групп может протекать по реакции с
гидроксильпыми группами:
РООН + РОН →РО•
+ Н2O + РО• (1.24)
По мере
окисления полимера такие группы накапливаются по реакции:
и в
результате распада гидропероксидных групп.
В растворе
кинетика автоокисления ПЭ подчиняется тем же закономерностям, что и окисление полимеров
в твердой фазе [13].
Инициированное
окисление полимеров осуществляется в лабораторных условиях; это позволяет
получить кинетические характеристики тормозящего действия ингибиторов. В
условиях переработки и эсплуатации протекает естественный процесс
автоокисления полимера. Его характерная особенность в том, что главным
инициатором цепного окисления полимера являются накапливающиеся в нем
гидропероксидные группы. Поэтому автоокисление любого органического соединения
RH протекает при переменной скорости инициирования. А так как скорость
образования гидропероксида зависит от количества и реакционной способности
введенного ингибитора, то возникает обратная связь: чем сильнее тормозящее
действие ингибитора, тем медленнее накапливается ROOH, меньше скорость
инициирования и длиннее период торможения.
Расходование
ингибитора при автоокислении RH носит нелинейный характер: по мере накопления
ROOH скорость расходования ингибитора возрастает. Так как гидропероксидные
группы распадаются, то возможны два режима ингибированного автоокисления:
нестационарный и квазистационарный по гидропероксиду [3].
В ходе
ингибированного автоокисления полимера протекают следующие процессы, которые
отражаются на кинетике расходования ингибитора и длительности торможения.
Во-первых,
накапливаются гидропероксидные группы, возрастает скорость инициирования и
вследствие этого ускоряется расходование ингибитора.
Во-вторых,
ингибитор улетучивается, кинетика испарения ингибитора в кинетическом режиме
описывается уравнением реакции первого порядка, в диффузионном режиме -
параболическим законом.
В-третьих,
продукты превращения ингибитора нередко сами обладают ингибирующим действием,
поэтому их накопление может замедлить расходование исходного ингибитора.
Поэтому кинетические кривые расходования ингибиторов при автоокислении
полимеров имеют самый разнообразный характер [1].
1.2 Влияние переходных металлов на окисление
полиолефинов
Поскольку полимеры получаются в промышленных
условиях, они неизбежно загрязняются примесями металлов в процессе получения.
Примеси металлов попадают в полимер или в виде
продуктов превращения металлсодержащих катализаторов (например, катализаторов
Циглера-Натта), или из-за контакта полимера с металлической аппаратурой. Кроме
того, при практическом использовании полимеров они часто приходят в контакт с
металлом, а это отражается на их стабильности [14].
Каталитическая активность металлов переменной
валентности связана с их склонностью участвовать в
окислительно-восстановительных реакциях и разрушать гидропероксид с
образованием свободных радикалов, например, по реакциям:
Men+ · ROOH ®
MeOHn+ + RO• (1.26)+ · ROOH ® Молекулярные
продукты
(1.27)
Обычно таким реакциям предшествует
комплексообразование (РООН входит во внутреннюю координационную сферу металла),
а распад РООН часто протекает по двум параллельным путям - гомолитическому и
гетеролитическому.
Преобладание гетеролитического распада приводит
в жидкости к низкой эффективности инициирования. В зависимости от того, какое
направление распада ROOH преобладает соединение металла ускоряет или замедляет
окисление. Ускорение окисления имеет место в тех случаях, когда эффективность
инициирования с катализатором выше, чем без катализатора. В противном случае
катализатор сокращает период индукции, но вместе с тем снижает и скорость
развившегося процесса [5].
Кинетика каталитического распада гидропероксида
зависит от предшествующих акту распада стадий комплексообразования, которые
протекают в режиме равновесных реакций. В некоторых случаях распад ROOH
протекает как реакция второго порядка по катализатору, что объясняют
образованием каталитически активных димеров.
В процессе окисления каталитический распад ROOH
осложняется следующими обстоятельствами [14].
Во-первых, высшая валентная форма катализатора
восстанавливается в низшую не только по реакции с гидропероксидом, но и по
реакциям с продуктами окисления: спиртами, кетонами, кислотами, альдегидами,
лабильными полифункциональными соединениями. В результате этого возрастает
скорость осуществления каталитического цикла:
+ → Men+1 → Men+ (1.28)
С другой стороны, эти кислородсодержащие
продукты конкурируют с ROOH как возможные лиганды во внутренней координационной
сфере металла, поэтому накопление кислородсодержащих продуктов снижает
концентрацию комплексов Men+∙ROOH и соответственно скорость генерирования
радикалов. Кроме того, появляются смешанные комплексы, такие, как ROH∙Men+∙ROOH,
активность которых часто ниже, чем комплексов Men+∙ROOH.
Во-вторых, в окисляющихся углеводородах
непрерывно генерируются пероксильные радикалы, которые, как и гидропероксид,
окисляют восстановленную форму катализатора:
++ RO2• → Men+1 + RO2• (1.29)
В таких условиях, когда эта реакция протекает
интенсивнее, чем генерирование радикалов, катализатор окисления превращается в
ингибитор. Это часто имеет место в начальный период, окисления, когда главный
источник радикалов - не каталитический распад РООН, а другие реакции, в которых
катализатор не участвует. В-третьих, в процессе окисления катализатор
постепенно переходит в малоактивное состояние из-за образования малоактивных
комплексов с продуктами окисления, в частности с водой, из-за превращения в
нерастворимые формы окислов и солей, которые выпадают в осадок [15].
В полимере (в твердом состоянии) спецификой
катализа является очень низкая скорость диффузии катализатора и ограниченная
подвижность гидропероксидных групп, связанных с макромолекулой. Из-за этих
обстоятельств катализ при окислении полимеров имеет локальный характер и
протекает только в тех микрообъемах, где катализатор и гидропероксидная группа
встречаются друг с другом. Скорость зарождения цепей в полимере тем выше, чем
выше содержание металла. Очевидно, сокращение периода индукции автоокисления
полимера в присутствии металлов связано с их инициирующим действием так же, как
и с каталитическим распадом гидропероксидных групп [16].
Оксиды меди, например, ускоряют окисление
полиэтилена: в их присутствии исчезает период индукции, окисление протекает с
более высокой скоростью.
По способности ускорять развившееся окисление
полимера, когда в нем накопились гидропероксидные группы, металлы располагаются
в следующий ряд: Со > Мn > Сu > Fe >V > Ni > Ti > А1 >
Mg > Ba [17].
1.3 Краткая характеристика ингибиторов окисления
Окисление полимеров, как и других органических
соединений, протекает как цепной процесс, в котором, чередуясь, участвуют
алкильные и пероксильные радикалы. Образовавшийся гидропероксид распадается на
радикалы и таким образом ускоряет окисление. Замедлить окисление можно, вызывая
интенсивный обрыв цепей по реакциям с пероксильными и алкильными радикалами или
вводя соединения, разрушающие гидропероксид без образования свободных радикалов
[6].
В некоторых системах одна молекула ингибитора
может обрывать большое число цепей (многократный обрыв цепей). Это наблюдается,
когда по реакции активного центра (R· или ROO·)
с продуктом превращения ингибитора (In·) снова образуется
исходный ингибитор или ингибитор действует каталитически, переводя активный ROO·
в малоактивный радикал. Стабильные нитроксильные радикалы (NO·)
многократно обрывают цепи при окислении полимеров, что лежит в основе высокой
эффективности стерически затрудненных аминов как светостабилизаторов.
Ингибиторы, обрывающие цепи, могут тормозить
двумя принципиально разными способами: либо они необратимо расходуются, обрывая
1-2 цепочки (стехиометрический обрыв цепей), либо вызывают обрыв большого числа
цепей (5-500), т. е. служат своеобразными катализаторами обрыва цепей. Наконец,
для торможения окисления часто используются смеси ингибиторов. В соответствии с
этим ингибиторы окисления можно разделить на следующие группы [15].
Ингибиторы, обрывающие цепи по реакции с
пероксильными радикалами. Такими ингибиторами являются ароматические соединения
со сравнительно слабыми связями О-Н и N-Н: фенолы, нафтолы, ароматические
амины, аминофенолы, диамины. Эти соединения обладают восстановительными
свойствами и быстро реагируют с пероксильными радикалами.
Ингибиторы, обрывающие цепи по реакции с
алкильными радикалами. К ним относятся соединения, быстро реагирующие с
алкильными радикалами: хиноны, иминохиноны, метиленхиноны, стабильные
нитроксильные радикалы, молекулярный йод. Алкильные радикалы быстро реагируют с
кислородом. Поэтому ингибиторы такого типа эффективны в условиях, когда
концентрация растворенного кислорода в окисляемом веществе низкая [11].
Ингибиторы, разрушающие гидропероксиды. Это
вещества, быстро реагирующие с гидропероксидами без образования свободных
радикалов: сульфиды, фосфиты, арсениты и т. д., а также тиофосфаты и карбаматы
металлов, разнообразные комплексы металлов.
Ингибиторы - дезактиваторы металлов. Соединение
металлов переменной валентности разрушают гидропероксиды с образованием
свободных радикалов, что ускоряет окисление. Такое катализированное окисление
удается замедлить, вводя комплексообразователь, который образует с металлом
комплекс, неактивный по отношению к гидропероксиду. В качестве ингибиторов
такого типа используются диамины, гидроксикислоты и другие бифункциональные
соединения, образующие с металлами прочные комплексы.
Ингибиторы многократного действия. При окислении
некоторых классов веществ (спирты, алифатические амины) образуются пероксильные
радикалы, обладающие как окислительным, так и восстановительным действием. В
таких системах ряд ингибиторов, обрывая цепи, снова регенерируется в актах
обрыва цепи: происходит каталитический обрыв цепей. Число обрывов цепей зависит
от соотношения скоростей реакций регенерации ингибитора и его необратимого
расходования. Многократный обрыв цепей наблюдается в ряде случаев и в
полимерах. Ингибиторами многократного обрыва цепей являются ароматические
амины, нитроксильные радикалы, соединения металлов переменной валентности.
Ингибиторы комбинированного действия. Некоторые
соединения тормозят окисление, одновременно вступая в несколько реакций.
Например, они реагируют и с алкильными, и с пероксильными радикалами (антрацен,
метиленхинон), разрушают гидропероксиды и обрывают цепи по реакции с РО2•
(карбаматы и тиофосфаты металлов). Такие соединения являются ингибиторами
комбинированного действия. В параллельные реакции может вступать одна и та же
группа Например, с двойной связью метиленхинона реагирует и Р• и РО2•. Часто в
молекуле имеются две или более функциональные группы, каждая из которых
вступает в соответствующую реакцию. Например, фенолсульфид реагирует с
гидропероксидом сульфидной группой и с РО2• фенольной группой. Наконец, в
разного типа реакции могут вступать исходный ингибитор и продукты его
превращения [8].
Синергические смеси ингибиторов. Часто для
торможения окисления используется смесь ингибиторов. Такая смесь эффективно
тормозит окисление, если ингибиторы взаимно усиливают действие друг друга, это
усиление называют синергизмом. Часто синергизм наблюдается для таких пар
ингибиторов, каждый из которых относится к ингибиторам разных типов. Например,
один ингибитор обрывает цепи, а другой разрушает гидропероксид. Однако
синергизм известен и для пар ингибиторов одного и того же типа. В практическом
отношении чрезвычайно интересно изучение явления синергизма, состоящего в том,
что стабилизирующее действие смеси двух веществ превышает сумму защитного
действия индивидуальных компонентов смеси [1].
Особое значение при стабилизации полимеров имеет
синергизм, который заключается в том, что защитное стабилизирующее действие
смеси веществ превышает аддитивный эффект защитного действия индивидуальных
компонентов (в концентрациях, равных суммарной концентрации всех
стабилизаторов). Синергизм определяют как превышение периода индукции окисления
в присутствии смеси веществ над периодом индукции в присутствии наиболее
эффективного компонента, взятого в концентрации, равной суммарной концентрации
смеси. Одно из важных условий синергизма - непрерывная регенерация
стабилизатора (ингибитора).
Синергизм - важный резерв повышения стабильности
полимерных материалов. Исследованию этого явления посвящено значительное
количество работ, большинство из которых направлено на эмпирический поиск и
подбор стабилизирующих композиций и смесей, способных давать синергический
эффект. Существует обратное явление - антагонизм АО. Например, многие
замещенные фенолы, а также некоторые амины в присутствии сажи (АО полиолефинов)
становятся менее эффективными [7].
Особое место занимает стабилизация полимеров от
действия света. Применяемые для этого светостабилизаторы поглощают
фотохимически активные компоненты солнечного света, дезактивируют возбужденные
молекулы, поглотившие квант света (тушение возбужденных состояний) или
замедляют темновые реакции, инициируемые светом. Применяют также
светостабилизаторы, дезактивирующие фотохимически активные примеси и продукты фотопревращений
[18].
Знание механизма стабилизации полимеров
позволяет прогнозировать и определять продолжительность надежной эксплуатации
полимерных материалов, правильно выбирать способы введения стабилизаторов.
Стабилизаторы можно вводить в полимеры на стадии их синтеза, переработки или в
готовое изделие.
Большинство полимерных материалов можно
стабилизировать различными способами. Но основным является внесение в
полиолефины различных стабилизирующих добавок.
Можно выделить три направления применения таких
добавок:
Стабилизация качества при обработке и
применении: антиоксиданты, термостабидизаторы и светостабилизаторы замедляющие
старение пластмасс при производстве или повышающие их рабочие характеристики.
Регулирование переработки: смазки,
разделительные средства или антиадгезивы предотвращающие нежелательные побочные
эффекты при производстве.
Придание новых свойств: антипирены, пигменты,
красители, антистатики или оптические отбеливатели модифицирующие различные
свойства конечных полимеров [19].
1.4 Химическая стабилизация структуры
полиолефинов
Основными принципами стабилизации
высокомолекулярных веществ являются структурно-физический и химический.
Суть структурно-физического принципа заключается
в воздействии физических факторов для создания молекулярной упаковки в
полимере, которая обеспечивает низкую молекулярную подвижность. Вследствие
этого происходит уменьшение химической реакционной способности макромолекул и
повышение стабильности материала. Именно этим объясняется повышенная
термоокислительная стабильность ориентированных полимеров по сравнению с
неориентированными [15].
Основной способ стабилизации - введение в
полимер специальных добавок (стабилизаторов или ингибиторов), замедляющих
старение. Роль стабилизаторов сводится либо к предотвращению образования
свободных радикалов, либо к взаимодействию молекул стабилизатора с растущими
радикалами и переводу их в неактивную форму.
Известно много веществ, используемых в качестве
стабилизаторов. В каждом конкретном случае при выборе катализатора учитывают
его эффективность, технологичность, стоимость и другие факторы. В зависимости
от назначения стабилизаторов различают антиоксиданты, светостабилизаторы,
антирады и др.
Стабилизаторы, применяемые для замедления
окислительной деструкции, называются антиоксидантами [8].
Существует огромное количество антиоксидантов.
Их можно классифицировать по химическому строению, по механизму ингибирования и
др. Ниже приведена их классификация по основному механизму ингибирования
процесса окисления. По этой классификации выделяют два типа ингибиторов [2].
К АО первого типа относятся замещенные фенолы и
многоядерные фенолы, в особенности бисфенолы, а также продукты конденсации
фенолов и комплексы с металлами, ароматические амины, аминофенолы и продукты
конденсации ароматических аминов. Основной механизм ингибирования окисления АО
первого типа заключается в отрыве углеводородным радикалом атома водорода от
молекулы АО (InH):
RO2 •+ InH ®
ROOH + In• (1.30)• + InH ® ROH + In• (1.31)
Активность радикала In• должна быть ниже
активности полимерных радикалов RO2•, RO•, с которым он взаимодействует - это
основное условие торможения окислительных процессов.
К АО второго типа относятся органические
серосодержащие соединения (тиоэфиры, тиобисфенолы, дисульфиды и др.),
органические фосфорсодержащие соединения, в особенности эфиры фосфористой и
производные тиофосфорных кислот. Эти соединения разрушают гидропероксиды, не
образуя радикалов. Сульфиды и дисульфиды реагируют с гидропероксидами, образуя
последовательно сульфоксиды и сульфоны, а фосфиты окисляются до фосфатов;
гидропероксид при этом восстанавливается до спирта. Недостаток этих АО - их
самоокисление [1].
1.5 Механизм влияния фенольных антиоксидантов на
полиолефины
Термоокислительная деструкция полимеров может
быть замедленна путём введения соответствующих стабилизаторов, называемых
антиоксидантами. Главным действующим началом фенольных АО, обеспечивающих их
способность тормозить окисление, является гидроксильная группа, присоединенная
к ароматическому ядру. Под фенольными АО понимают любые соединения вида
Ar(OH)n. В зависимости от природы ароматического фрагмента различают
собственные фенолы, а также нафтолы, оксипроизводные других конденсированных
ароматических углеводородов и гетероароматических соединений [20].
В зависимости от числа OH-групп, присоединенных
к одному ароматическому ряду, различают монофенолы и полифенолы.
В зависимости от числа фрагментов Ar(OH)n в
молекуле АО говорят о моноядерных и полиядерных фенолах( бисфенолы, трисфенолы
и т.д.).
Взаимодействие с перекисными радикалами
окисляющегося субстрата - принципиальная реакция, которая прежде всего
определяет способность фенольных соединений тормозить цепное окисление.
Продуктом реакции является феноксильный радикал:
ОН + RO2 → ArО• + RООН. (1.30)
Феноксильные радикалы образуются при
взаимодействии с фенолами и других радикалов: алкоксильных, алкильных,
карбоксильных, а также синглетного кислорода, молекулярного кислорода,
триплетных состояний карбонильных соединений, одноэлектронных окислителей
РbO2,Аg2O, [Fе(CN)6]3- и т.д.[21].
Нужно отметить еще две реакции с участием
фенолов которые могут протекать в условиях ингибированного окисления:
взаимодействие с RООН и O2. Реакция ArОН с RООН происходит следующим образом
[22]:
ОН + RООН → RО• + Н2O + ArО• (1.31)
Образующиеся в этой реакции алкоксильные
радикалы частично выходят в объем и могут служить дополнительным источником
инициирования.
Практически все фенольные антиоксиданты при
длительном хранении на воздухе медленно окисляются как в кристаллическом
состоянии, так и в растворах. Формально начальную стадию взаимодействия ArОН с
O2 обычно описывают реакцией
ОН + O2 → ArО• + НO2• (1.32)
Как уже говорилось, не существу, единственным
первичным продуктом превращения фенольногс антиоксиданта являются феноксильные
радикалы. От направления их дальнейших реакций зависит как набор молекулярных
продуктов, так и в значительной степени эффективность антиоксиданта [23].
О + RO2 → RO2ArО• (QP) ( 1.33)
В условиях ингибированного окисления с реакцией
(1.33) конкурируют
процессы бимолекулярной гибели ArО•: О• + ArО• →
продукты (1.34)
Их направление зависит от структуры ArО. Для
феноксильных радикалов, у которых хотя бы одно из "активных"
положений 2,4 или 6 не замещено, характерна рекомбинация с образованием
димерных фенольных соединений.
Рассмотрим далее реакции феноксильных радикалов
с молекулами. Наибольшее значение имеют следующие реакции радикального
замещения:
О + RООН → ArОН + RO2, (1.35)О + RН →
ArОН + R, (1.36)О + ArОН• → ArОН + ArO•. (1.37)
В ряде практически важных случаев равновесие
довольно сильно смещено в сторону образования RO2. При этом, как легко понять,
антиоксидант не может эффективно тормозить окисление.
С точки зрения способности тормозить окисление
крайне нежелательна также реакция (1.36), в которой феноксильный радикал
атакует связь С-Н в молекуле окисляемого субстрата. "Выменивание" в
этой реакции химически сравнительно инертного феноксила на активный радикал R
алкильного типа приводит к регенерации цепи [17].
Обменные реакции (1.37) представляют для нас
интерес с нескольких точек зрения. Во-первых, стабилизирующие композиции в
некоторых случаях включают два фенольных соединения. Во вторых, фенольные
соединения, отличные от исходного антиоксиданта, образуются уже в ходе
ингибированного окисления. Наконец, изучение равновесия (1.37) имеет
определенное методическое значение, поскольку позволяет оценивать прочности
связи О-Н в фенолах, а также реакционную способность фенолов и феноксильных
радикалов [12].
Процессы присоединения ArО к ненасыщенным
соединениям не играют, по-видимому, такой важной роли, как реакции замещения.
Их механизм и особенно кинетика довольно мало изучены. Типичным, вероятно,
является механизм, приводящий к образованию циклогексанодиенов.
Особое место занимает реакция феноксильных
радикалов с молекулярным кислородом. В результате этого процесса образуются
симметричные хинолидные перекиси [18].
В результате окислительных превращений фенольных
соединений возникает довольно разнообразный набор продуктов, многие из которых
принимают участие в дальнейших реакциях. Главный канал образования хинонов -
распад хинолидных перекисей. Кроме того, хиноидные соединения могут возникать в
результате диссоциации алкоксизамещенных феноксилов, а также при
диспропорционировании оксизамещенных феноксильных радикалов. Возможный вклад
хинонов в кинетику ингибированного окисления связан с их способностью
эффективно присоединять алкильные радикалы
Отметим, что сами фенольные антиоксиданты
малоактивны по отношению к алкильным радикалам и хиноны - по существу,
единственный продукт (за исключением, быть может, метиленхинонов.), способный
обрывать цепи окисления не только в результате реакции с RO2,но и при
взаимодействии с алкильными радикалами. Это может иметь важное значение,
например, в условиях дефицита О2.
Существует несколько путей возникновения в ходе
ингибированного окисления фенольных соединений, отличных от исходного
антиоксиданта, Отметим наиболее важные:
димеризация частично замещенных феноксильных
радикалов;
диспропорционирование алкилированных феноксилов,
приводящее
к образованию метиленхинонов, с последующей
димеризацией последних;
последовательное вовлечение в реакцию ОН-групп
многоядерных фенолов;
взаимодействие алкильных радикалов с хинонами и
метиленхинонами и превращение образующихся феноксильных радикалов в фенолы,
например в результате обменных реакций с исходным фенолом.
Не вызывает сомнений, что вторичные фенольные
соединения наряду с исходным антиксидантом обрывают цепи окисления и принимают
участие в других реакциях. Это значительно усложняет механизм ингибированного
окисления и его кинетический анализ [8].
2. Программа, объект и методика исследования
.1 Программа исследований
Опыты проводились в МННЛ «Физика и химия
полимеров» в период июнь-ноябрь 2011 года и июнь-ноябрь 2012 года.
Для достижения поставленной цели (изучение
изменений эффективности фенольных антиоксидантов в условиях окисления
полиолефинов, контактирующих с переходными металлами разной валентности) было
определена программа исследований:
изучение научной литературы по основным
закономерностям процесса окисления (старения) полимеров;
изучение научной литературы по влиянию металлов
(и их оксидов) на процессы окисления ингибированного полиэтилена;
подготовка полимерных композиций на основе
полиэтилена, фенольного и аминного антиоксидантов и оксидов металлов; расчёт
навесок для композиций;
изготовление полимерных образцов методом
термического прессования;
проведение термоиспытаний полученных полимерных
образцов;
контроль структуры полиэтилена при окислении
методом ИК-спектроскопии;
обсуждение результатов эксперимента,
формулирование основных выводов и практических рекомендаций.
2.2 Характеристика основных объектов
исследования
Полиэтилен низкого давления (ПЭНД). Полиэтилен
(-СН2-СН2-)n - карбоцепной термопластичный кристаллический полимер белого цвета
со степенью кристалличности при 20°С 0,5-0,9 [соколов]. Свойства ПЭНД
представлены в таблице 1. ПЭНД получают в реакторах-автоклавах колонного типа
при давлении 0,25-0,5 МПа и температуре не выше 80 0С.[26].
Таблица 1 - Свойства полиэтилена
|
Показатель
|
ПЭНД
|
|
Молекулярная
масса, тыс. единиц Плотность, т/м3 Степень кристалличности Температура
плавления, °С Температура хрупкости, °С Уд. электрическое сопротивление, Ом-м
|
50-800
0,95-0,96 0,75-0,85 120-125 100-150 1015
|
ПЭНД получается координационно-ионной
полимеризацией этилена в растворе бензина при температуре 70-80°С и давлении
0,15-0,3 МПа в присутствии комплексных металлоорганических катализаторов
(катализаторов Циглера-Натта). Среди них наибольшее распространение получили
катализаторы состава Al(C2H5)2Cl-TiCl4.
Технологический процесс производства ПЭНД
включает следующие основные стадии: приготовление катализаторного комплекса,
полимеризация этилена, выделение порошкообразного полимера и разложение
остатков катализатора, промывка и сушка полиэтилена.
Скорость полимеризации и свойства получаемого
ПЭНД зависит от температуры, давления и активности катализатора, которая
определяется мольным соотношением диалкилалюминия и тетрахлорида титана. При
повышении содержания последнего в контактной массе возрастает скорость процесса
и выход ПЭ, но снижается его молекулярная масса. Для регулирования молекулярной
массы полимера в этилен вводится водород, который играет роль передатчика цепи.
Катализаторный комплекс легко разрушается под воздействием кислорода воздуха и
влаги. Поэтому процесс полимеризации проводится в атмосфере азота и в среде
обезвоженного бензина [26].
Ирганокс 1010. В качестве добавок, ингибирующих
окисление, в работе применялись: ирганокс 1010
(3,5-ди-третбутил-4-окси-фенилпропионовой кислота) - АО фенольного типа [27].
Ирганокс представляет собой белый или кремовый
кристаллический порошок. Нерастворим в воде. Раздражающим действием не
обладает. Используется в качестве термостабилизатора. Антиоксидант общего
назначения, эффективно защищает полимеры от теплового старения, повышает
сопротивление резин разрушению при многократных деформациях. Применяется в
качестве стабилизатора синтетических каучуков и полиолефинов, а также для
защиты резин на основе каучуков общего назначения [24].
Нафтам-2 или неозон Д - порошок от
светло-коричневого до серого цвета, применяется в качестве стабилизатора
синтетических каучуков и для защиты резин на основе каучуков общего назначения.
Широко используется в производстве резиновых технических изделий. Химическое
название нафтама-2 (неозона Д): фенил-β-нафтиламин.
Легко растворяется в бензоле, толуоле, хлороформе, ацетоне, этилацетате;
растворим в спирте и четыреххлористом углероде. Нерастворим в воде.
Растворяется в каучуке в количестве 115 вес.%. Антиоксидант общего назначения,
эффективно защищает каучуки и резины от теплового старения, повышает
сопротивление резин разрушению при многократных деформациях. Выцветание
наблюдается в резинах из НК при введении более 1,5 вес. ч., в резинах из
хлоропренового каучука при введении более 3 вес. ч. и в резинах из
бутадиен-стирольного каучука - более 2 вес. ч. На свету сильно окрашивает
резину и контактирующие с ней материалы. Применяется в качестве стабилизатора
синтетических каучуков и полиолефинов, а также для защиты резин на основе
каучуков общего назначения. Широко используется в производстве резиновых
технических изделий, подвергающихся тепловому старению и динамическим
деформациям. Не может применяться для изготовления изделий светлых,
медицинского назначения и находящихся в контакте с пищевыми продуктами.
Эффективность повышается при добавлении других антиоксидантов, например
N,N'-замещенных n-фенилендиамина [22].
Получение. Неозон Д получают следующим образом.
В стальной эмалированный реактор загружают приготовленную в другом аппарате
смесь анилина и 2-нафтола, добавляют небольшое количество солянокислого анилина
и при размешивании нагревают реакционную массу. Реактор оборудован двумя
последовательно соединенными холодильниками - обратным и прямым. Пары воды,
образующейся в ходе реакции, и пары анилина сначала поступают в обратный
холодильник, охлаждаемый горячей водой. Здесь анилин конденсируется и стекает в
реактор, а пары воды проходят через обратный холодильник и поступают затем в
прямой холодильник, где конденсируются и стекают в приемник. Таким образом
непрерывно удаляется из реакционной массы вода, образующаяся в ходе реакции.
Температуру реакционной массы постепенно повышают до 250-260 С0. По окончании
араминирования (определяют по анализу пробы реакционной массы) добавляют щелочь
для нейтрализации кислоты и отгоняют с водяным паром избыточный анилин. После
удаления анилина реакционную массу упаривают и расплавленный неозон Д передают
на очистку и кристаллизацию.
Применение. Применяется в качестве стабилизатора
синтетических каучуков и для защиты резин на основе каучуков общего назначения.
Широко используется в производстве резиновых технических изделий, подвергающихся
тепловому старению и динамическим деформациям. Не может применяться для
изготовления изделий светлых, медицинского назначения и находящихся в контакте
с пищевыми продуктами. Эффективность повышается при добавлении других
антиоксидантов, например N,N'-замещенных n-фенилендиамина [24 рогицын].
Свойства. Неозон Д представляет собой белый
порошок с температурами плавления 104-108 С0, кипения 395,5 С0. Растворяется в
бензоле, горячем спирте, эфире, ацетоне, нерастворим в воде, малолетуч.
Представляет собой тонкоизмельченный порошок светло-серого или
светло-коричневого цвета.
Неозон Д - важнейший противостаритель для
каучуков и резин. Небольшая его добавка значительно удлиняет срок службы
резиновых изделий.
При длительном хранении неозон Д слеживается в
монолитную массу, которую трудно ввести в полимер. Для устранения этого
недостатка в него вводят небольшие добавки веществ, препятствующих слипанию и
облегчающих распределение стабилизатора в полимере.
Для ингибирования процесса термоокислительной
деструкции достаточно добавления 0,1-0,25 % антиоксиданта. Увеличение
количества добавляемого антиоксиданта не дает существенных результатов. Поэтому
для уменьшения интенсивности термоокислительной деструкции полиолефина в
процессе формования волокна при повышенных температурах в состав полимера
вводят небольшое количество антиоксиданта [22].
Оксиды металлов:
Со2О3 - черные гексагональные мелкие кристаллы.
Плотность равна 5,34 г/см3. Устойчив в виде моногидрата. Превращается в Со3О4
при 265оС, в СоО при 940оС с выделением кислорода. Окисляет хлороводородную
кислоту с выделением хлора. Восстанавливается водородом или метаном. Получают
дегидратацией Со2О3.nН2О или прокаливанием нитрата кобальта (II) при 180оС.
Применяют как пигмент для эмалей и глазурей.
Со3О4 - парамагнитные черные октаэдрические
кристаллы со структурой шпинели. Плотность равна 6,1-6,2 г/см3. Превращается в
СоО при 940оС с выделением кислорода. Восстанавливается водородом, углеродом,
алюминием. Растворяется в кислотах и расплавленных щелочах. Получают
нагреванием порошкообразного металлического кобальта на воздухе при 300-400оС,
нагреванием на воздухе СоО или Со(ОН)2 при температуре выше 100оС. Применяют
для изготовления стекла, сильно поглощающего ультрафиолетовые лучи, а также в
качестве катализатора.
Cr2O3 -
очень твёрдый тугоплавкий порошок зелёного цвета. Температура плавления 2435
°C, кипения около 4000 C. Плотность 5,21 г/см³. Нерастворим
в воде. По твердости близок к корунду
<#"605275.files/image002.gif">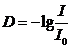 , (2.1)
, (2.1)
где I -
интенсивность пропускания света в области полосы поглощения 1720 см-1;- фоновое
значение интенсивности света.
В качестве
аналитической для проведения количественного анализа используют полосу
поглощения при 1720см-1 (5,82 мкм), соответствующую валентным колебаниям
карбонильных групп кетонного и альдегидного типа. В качестве внутреннего
стандарта выбирают полосу при 1470 см-1, вызванную деформационными колебаниями
СН3- и СН2-групп, остающуюся без изменения. Степень окисления полимера при
заданном времени, стабилизаторе и температуре оценивают по отношению оптических
плотностей в точках 1720 и 1470см-1 [31].
К числу
основных преимуществ ИК-Фурье спектрометров можно отнести высокую
чувствительность, которая позволяет регистрировать предельно низкие
концентрации, а так же малые количества вещества; время, нобходимое для
получения спектра составляет 2-20 с, благодаря чему ИК-Фурье спектрометр
позволяет выполнять экспресс-измерения, перейти от выборочного контроля
продукции к сплошному, контролировать параметры технологических процессов в
реальном времени; способствует повышению надежности измерений, позволяет
автоматизировать учет результатов, а так же повысить эффективность их
обработки; интерферометр не требует настройки, имеется встроенный стандарт
длины волны, процесс тестирования и поверки автоматизирован; спектрометр может
быть легко адаптирован для решения специализированных задач; управляется с
помощью персонального компьютера, программное обеспечение работает в среде
Windows 9x/XP/Vista/7, обеспечивает измерение спектров, тестирование
спектрометра, работу со спектральной базой данных, разработаны пакеты программ
для решения различных прикладных задач: многокомпонентный количественный
анализ, факторный анализ, идентификация спектров по заданной библиотеке и
прочее [24].
В основе
анализа лежит связь инфракрасного спектра поглощения и состава образца.
Местоположение полос в спектре поглощения несет информацию о качественном
составе образцов, а интенсивность полос - о концентрации соответствующего
компонента. Для количественного анализа образца необходимо знать зависимость
между интенсивностью поглощения и концентрацией компонента или свойством
образца.
Предварительное
определение зависимости между показателем поглощения (пропускания) и
концентрацией компонента или свойством образца называется калибровкой. Под
проведением калибровки понимают регистрацию спектров партии образцов с
известными концентрациями компонентов или известными свойствами. По этим данным
рассчитывается калибровочная модель, которая связывает содержание определяемого
компонента с результатом спектрального анализа и позволяет по спектру
поглощения количественно определить интересующий компонент.
Для
проведения калибровки отбирается набор образцов, представительный к тем
образцам, которые будут в дальнейшем анализироваться. Калибровочный набор
включает образцы, свойства которых охватывают весь диапазон возможных значений
определяемых компонентов и свойств анализируемых образцов. Калибровочные
образцы анализируются стандартными химическими (референтными) методами для
определения в них концентраций компонентов или свойств [31].
Производится
регистрация спектров образцов калибровочного набора и рассчитывается
калибровочная модель, связывающая спектральные данные со свойствами образца.
Для расчета модели используются методы мультивариантной математики, например,
метод регрессии по основным компонентам, метод дробных наименьших квадратов,
множественная линейная регрессия [25].
Определенная
калибровочная модель применяется для анализа образцов, свойства которых
укладываются в диапазон значений концентраций и свойств обучающего набора.
Погрешность анализа зависит от погрешности спектрального анализа и погрешности,
связанной с построением калибровочной модели.
ИК спектры
поглощения, отражения или рассеяния несут чрезвычайно богатую информацию о
составе и свойствах пробы. Сопоставляя ИК спектр образца со спектрами известных
веществ, можно идентифицировать неизвестное вещество, определить его основной
состав, обнаружить примеси, провести анализ. Современные спектрометры
определяют ИК спектр сканированием по сдвигу фаз между двумя частями
разделенного светового пучка (Фурье-спектрометрия). Этот метод дает
значительный выигрыш в фотометрической точности и точности отсчета длины волны
[35].
Анализ
структуры полиэтилена на основании его колебательного спектра является классическим
примером достижений ИК-спектроскопии полимеров. Структура полиэтилена стала
объектом многочисленных работ как теоретических, так и экспериментальных. Это
обусловлено, во-первых, простотой ИК-спектра полиэтилена, во-вторых,
возможностью сравнения его спектра со спектрами длинноцепочечных нормальных
парафинов. Большое техническое значение полиэтилена также явилось причиной
широкого исследования особенностей строения этого полимера (определение двойных
связей в основной цепи, степени разветвленности, кристалличности, ориентации и
деструкции) [20].
Поглощение
ИК-излучения связано с колебаниями молекулы и соответственно ее групп.
Взаимодействие между группами оказывает влияние на колебательные частоты. В
низкомолекулярных соединениях, имеющих кристаллическую структуру, существует
однозначная связь между колебательным спектром этого соединения, его структурой
и силами межмолекулярного взаимодействия. Правда, данная макромолекула в
определенных условиях имеет определенный колебательный спектр. Но этот спектр в
большей мере, чем для низкомолекулярных соединений, зависит от физических
воздействий, изменяющих конформацию макромолекул и надмолекулярную структуру. В
колебательном спектре такие изменения находят отражение, и это дает ценную
информацию о структуре полимера, что в свою очередь способствует решению ряда
физико-химических проблем [36].
ИК-спектры полиэтилена (рисунок 2) представляют
собой сумму спектров аморфной и кристаллической фаз полимера. В кристалле цепи
полиэтилена имеют плоскую зигзагообразную конформацию. В аморфной же фазе цепи
содержат статистический набор неплоскостных поворотных изомеров (гош-формы),
которые частично переходят в плоскостную (транс-форму) только при растяжении
образца. Возникновение новых конформаций ограничено лишь постоянством значений
валентных углов и длин связей [19].
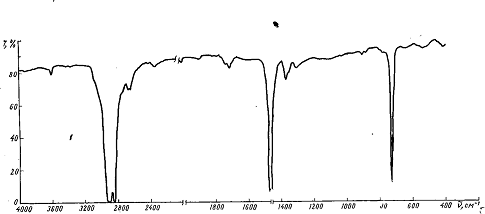
Рисунок 2 - ИК-спектр полиэтилена низкого
давления
Основные характеристики ИК-спектров полиэтилена
могут быть выведены из анализа колебаний изолированной цепи. Полная интерпретация
спектра требует учета реальной кристаллической структуры полимера. В
технических полимерах в зависимости от способа получения их (полиэтилен
высокого давления, полиэтилен низкого давления) наряду с метиленовыми звеньями
присутствуют также и различные моноалкилэгиленовые звенья. Они, как и
ненасыщенные группы, образуют разветвления, которые обуславливают
дополнительные полосы поглощения в спектре. В расчетах, проведенных для
бесконечно длинной полиэтиленовой цепи, влияние разветвлений на спектр
полиэтилена, как правило, не рассматривается. Повторяющейся единицей в
бесконечно длинной изолированной цепи полиэтилена являются две СН2-группы [21].
3. Результаты исследований и их обсуждение
.1 Окисление полиэтилена, содержащего фенольный
антиоксидант
Первые результаты исследований окисления
ингибированного фенольным антиоксидантом полиэтилена в контакте с активной
медной подложкой были выполнены в 1970-х годах. Именно в работах тех лет было
показано, что контакт полиэтиленовых пленок с медной подложкой ускоряет расход
фенольного антиоксиданта, и при достижении достаточно низкой концентрации
модификатора начинается автокаталитическое окисление полимера.
На рисунке 3 приведены данные по накоплению
карбонильных групп в пленках полиэтилена, ингибированных ирганоксом 1010 и
окисляемых на подложках из меди и цинка [37].
Результат эксперимента оказался неожиданным -
если медная подложка ускоряет окисление ингибированного полимера, то цинковая,
наоборот, выступает в роли дополнительного ингибитора окислительного процесса.
В результате ИПО полиэтиленовых пленок, окисленных на цинке, оказался выше, чем
у пленок, контактирующих при окислении с пассивной (KBr) подложкой (рисунок 3,
кривые 1-3).
Неожиданность в поведении цинка заключается в
том, что в случае контакта этого металла с неингибированным полиэтиленом, он
выступает в роли катализатора окисления. Однако при введении в полиэтилен
фенольного антиоксиданта, контакт полимера с цинковой подложкой приводит не к
сокращению ИПО, а, наоборот, к его увеличению [38].
Так для пленок толщиной 100 мкм ИПО
ингибированного полиэтилена на цинке составляет 74 часа (рисунок 3, кривая 3),
в то время как для таких же плёнок, окисляемых на пассивной подложке, он
оказался равным 31 часу (рисунок 3, кривая 3, рисунок 3, кривая 1).

Рисунок 3 -
Зависимость оптической плотности 1720 см-1 в ИК-спектрах полиэтиленовых пленок,
содержащих 0,1% мас. ирганокса 1010, от продолжительности их окисления при
температуре 150 °С на
пластинах из KBr (1), цинковой (2, 3) и медной (4, 5) подложках. Толщина пленок
100 мкм (1, 3, 4) и 70 мкм (2, 5)
Эффект дополнительного ингибирования
окислительного процесса в пленках на цинке зависит от толщины пленки. Можно
заметить в общем плане - с уменьшением толщины пленки эффективность влияния
подложки усиливается. Это относится к обоим исследуемым металлам. В случае
меди, чем тоньше пленка, тем в большей мере сокращается ИПО (рисунок 3, кривые
4, 5), для цинковой подложки картина обратная - с уменьшением толщины пленки
ИПО увеличивается (рисунок 3, кривые 2, 3).
Установленный характер влияния металлических
подложек на окисление полиэтилена, ингибированного играноксом 1010, был
подтверждён для другого вида образцов - ингибированного полиэтилена,
наполненного этими металлами [39].

Рисунок 4 - Зависимость оптическойплотности 1720
см-1 в ИК-спектрах полиэтиленовых пленок, содержащих 0,1 % мас. ирганокса 1010
и 10 (2); 15 (3) % мас. медного или 10 (4); 15 (5) % мас. цинкового порошка, от
времени окисления пленок при температуре 150 °С на пластинах из KBr. Толщина пленок
100 мкм [189]
Как видно из рисунка 4, при введении в полимер
порошка цинка ИПО увеличивается (кривые 1, 2, 3), а при введении порошка меди
уменьшается (рисунок 4, кривые 1, 4, 5). Чем выше концентрация наполнителей,
тем сильнее проявляется эффект изменения ИПО.
В обоих рассмотренных случаях: при наполнении
ингибированного полимера порошком цинка или меди (окисление плёнок ведется на
подложках из KBr), так и при уменьшении толщины пленки покрытия на
металлической подложке (плёнка полимера содержит только антиоксидант)
увеличивается величина площади поверхности металлической подложки, приходящейся
на единицу массы полимера. Как следствие, возрастает эффект, оказываемый
металлом на окислительный процесс [40].
Очевидно, что к аналогичному результату можно придти,
не изменяя массовую (объемную) концентрацию наполнителя в полимере, а только
лишь уменьшая размеры его частиц, то есть переходя к более мелким частицам
наполнителя. При этом также возрастает общая поверхность контакта полимера с
металлом и, как следствие, увеличивается эффект, оказываемый металлом на ход
окислительного процесса [39].

Рис. Зависимость оптической плотности полосы
поглощения 1720 см-1 в ИК-спектрах плёнок ПЭ, содержащих 0,1 % Д (2-4), от
продолжительности их окисления на медной (3) и KBr (1, 2, 4) подложках. Кривая
4 - пленка содержат 10% масс. дисперсной меди. Температура окисления 150°С.
Активность металлов к окислению ингибированного
полимера удобно характеризовать с помощью показателя относительной активности τ,
равного отношению ИПО полимера, окисляемого соответственно в контакте с
пассивной и активной подложками (τ=ИПОпас/ИПОакт).
Этот показатель в равной степени можно рассчитывать как для образцов, в которых
исследуемый металл используется либо в виде наполнителя полимера, либо в виде
подложки для пленки ингибированного полимера.
Если металл активизирует окислительный процесс и
ИПО сокращается, то значение показателя относительной активности будет больше
единицы. Если металлическая подложка, наоборот, способствует увеличению
эффективности используемого антиоксиданта и ИПО увеличивается (как в случае
контакта цинка с полиэтиленом, ингибированным фенольным антиоксидантом), то
рассчитываемый показатель уже будет меньше единицы.
Понятно, что при сопоставительных исследованиях
нужно использовать значения показателей, рассчитанное для образцов одного вида,
размеров и окисленных в одних и тех же термических условиях. Примеры
использования показателя относительной активности металлических подложек содержаться
в работах авторов [37].
3.2 Влияние валентности металла оксида на
окисление полиэтилена, ингибированного фенольным антиоксидантом
По результатам проведенных исследований можно
выделить две группы добавок: одна группа добавок при увеличении валентности
металла оксида в составе композиции приводила к сокращению ИПО ингибированной
полимерной пленки; другая группа, наоборот, при увеличении валентности металла
оксида приводила к увеличению продолжительности ИПО.
В таблице 2 и 3 представлены данные по кинетике
изменения оптической плотности полосы 1720 см-1 в образцах полиэтиленовых
плёнок, содержащих 0,1% ирганокса 1010 и 1 % оксида металла, окисленных при
температуре 150° С
Таблица 2 - Изменение оптической плотности
полосы 1720 см-1 (D1720/1470) первой группы добавок
|
Добавка-оксид
|
Co2O3
|
Co3O4
|
Cr2O3
|
CrO3
|
|
D
1720/1470
|
9ч-0,004±0,001
10ч-0,010±0,011 13ч-0,0120±0,012 16ч-0,0150±0,012 19ч-0,150±0,011
20ч-0,020±0,001 23ч-0,530±0,012 27ч-4,700±0,012
|
10ч-0,01±0,01
13ч-0,01±0,03 16ч-0,01±0,07 19ч-0,01±0, 01 20ч-0,02±0,2 23ч-0,06±0, 01
27ч-1,86±0, 01
|
23ч-0,01±0,06
27ч-0,02±0,05 28ч-0,02±0,03 29ч-0,03±0,01 30ч-0,05±0,03 31ч-0,09±0,02
32ч-0,20±0,05 33ч-0,34±0,07 35ч-0,86±0,04 36ч-1,25±0,02
|
0ч-0,00±0
1ч-0,72±0,05
|
|
ИПО
|
20
|
22
|
30
|
0,1
|
Таблица 3 - Изменение оптической плотности
полосы 1720 см-1 (D1720/1470) второй группы добавок
|
Добавка-оксид
|
Fe2O3
|
FeO
|
Sb2O3
|
Sb2O5
|
|
D
1720/1470
|
18ч-0,02±0,02
20ч-0,03±0,05
|
3ч-0,03±0,02
8ч-0,04±0,01 10ч-0,07±0,05 11ч-0,07±0,01 13ч-0,12±0,05 14ч-0,16±0,07
15ч-0,14±0,08 17ч-0,20±0,05 18ч-0,38±0,09 20ч-0,44±0,01
|
1ч-0,0±0,0
3ч-2,7±0,3
|
20ч-0,01±0,07
23ч-0,09±0,03 27ч-1,38±0,02
|
|
ИПО
|
20
|
8,5
|
1
|
22
|
К первой группе добавок относятся оксиды
кобальта (рис.5, кривые 1 и 2), оксиды хрома (рис.6, кривые 1 и 2), ко второй
группе - оксиды железа (рис.7, кривые 1 и 2) и сурьмы (рис.8, кривые 1 и 2).
Необходимо отметить, что при введении добавок
(оксидов металлов) все они снижают продолжительность ИПО (рис.5-6). ИПО
ингибированного ПЭ без добавок составляет 31 час (рис.3, кривая 1). Минимально
изменяет ИПО оксид хрома (III) (рис.6, кривая 1).
Рассмотрим влияние добавок первой группы. В
общем случае с ростом валентности возрастают кислотные свойства оксида. При
этом если сравнить оксиды кобальта (IV), то при изменении валентности на 1
основные свойства изменяются до амфотерных (данные по Лурье). Такое небольшое
изменение валентности приводит к небольшому сокращению ИПО всего на 2 часа
(рис.5, кривые 1 и 2).
Оксид хрома (III) и оксид хрома (VI) отличаются
на 3 единицы валентности и существенно отличаются по свойствам: оксид хрома
(VI) обладает очень яркими окислительными свойствами, а оксид хрома (III) -
амфотерными [41]. И как следствие мы видим существенное сокращение ИПО при
переходе от оксида хрома (III) к оксиду хрома (VI) - от 30 часов до нескольких
минут (рис.6, кривые 1 и 2)

Рисунок 5 - Изменение оптической плотности 1720
см-1 при окислении пленок толщиной 100 мкм, содержащих 0,1% ирганокса 1010
(1,2) и 1% Co2O3(1) и 1%Co3O4(2)

Рисунок 6 -
Изменение оптической плотности 1720 см-1 при окислении пленок толщиной 100 мкм,
содержащих 0,1% ирганокса 1010 (1,2) и 1% CrO3(1) и 1%Cr2O3(2)
Данные
таблицы 3 показывают, что возрастание валентности металла может приводить и к
возрастанию ИПО полимера, ингибированного ирганоксом 1010. К этой группе
добавок относятся оксиды железа (рис.7, кривые 1 и 2)и оксиды сурьмы (рис.8,
кривые 1 и 2).
Рассмотрим
влияние добавок второй группы. Также как и в первой группе с ростом валентности
возрастают кислотные свойства оксида, но в этом случае усиление кислотных
свойств приводит к увеличению ИПО, в отличие от влияния первой группы добавок.
При этом если сравнить оксиды железа (II), то при изменении валентности на 2
основные свойства изменяются до амфотерных [42]. Такое изменение валентности
приводит к увеличению ИПО всего на 11.5 часов (рис.7, кривые1 и 2).
Оксид сурьмы
(III) и оксид сурьмы (V) отличаются на 2 единицы валентности и существенно
отличаются по свойствам: оксид сурьмы (V) обладает очень яркими окислительными
свойствами, а оксид сурьмы (III) - амфотерными. [41]. И как следствие мы видим
существенное сокращение ИПО при переходе от оксида сурьмы (III) к оксиду сурьмы
(V) - от 22 часов до 1 часа (рис.8, кривые 1 и 2).
Поведение
оксидов железа и оксидов сурьмы объясняется тем, что как FeO, так и Sb2O3
является неустойчивыми оксидами и при окислении переходят в F2O3 и Sb2O5
соответственно, которые дальше подвергаются окислению.
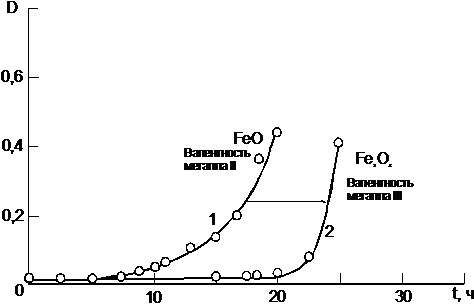
Рисунок 7 -
Изменение оптической плотности 1720 см-1 при окислении пленок толщиной 100 мкм,
содержащих 0,1% ирганокса 1010 (1,2) и 1% FeO(1) и 1%Fe2O3(2)

Таким образом, в результате исследований было
установлено, что оксиды металлов с переменными валентностями оказывают
различное влияние на процесс окисление полиэтилена, ингибированного ирганоксом
1010. Было выделено две группы металлов: одна группа металлов при увеличении
валентности металла оксида в составе композиции приводило к сокращению ИПО
ингибированной полимерной пленки, другая группа металлов при увеличении валентности
металла оксида приводила, наоборот, к увеличению продолжительности ИПО. К
первой группе относятся оксиды кобальта и хрома. Ко второй группе относятся
оксиды железа и сурьмы.
Результаты исследований опубликованы в
материалах Международной научно-технической конференции „Технология-2012” [43].
Заключение
Экспериментально установлено, что контакт с
металлическим цинком полиэтилена, ингибированного фенольным антиоксидантом -
ирганоксом 1010, приводит к увеличению термоокислительной стойкости полимера.
Предположено, что увеличение эффективности фенольного антиоксиданта в
присутствии цинка связывается с регенерацией отработанного антиоксиданта на
металле. В результате антиоксидант получает возможность многократно участвовать
в реакциях обрыва окислительных цепей, благодаря чему индукционный период
окисления полимера увеличивается.
По результатам исследований было установлено,
что оксиды металлов с переменными валентностями оказывают различное влияние на
процесс окисление полиэтилена, ингибированного ирганоксом 1010. Было выделено
две группы металлов: одна группа металлов при увеличении валентности металла
оксида в составе композиции сокращает ИПО ингибированной полимерной пленки
(оксиды кобальта и оксиды хрома), другая группа металлов при увеличении валентности
металла оксида, наоборот, увеличивает продолжительность ИПО полимера (оксиды
железа и оксиды сурьмы).
Список использованных источников
Денисов,
Е.Т. Окисление и деструкция карбоцепных полимеров / Е.Т. Денисов. - Л.: Химия,
1990. - 288 с.
Кирилова,
Э.И. Старение и стабилизация термопластов / Э.И. Кирилова, Э.С. Шульгина. - Л.:
Химия, 1988. - 240 с.
Эмануэль,
Н.М. Химическая физика старения и стабилизации полимеров / Н.М. Эмануэль, А.Л.
Бучаченко. - М.: Наука, 1988. - 368 с.
Гладышев,
Г.П. Стабилизация термостойких полимеров / Г.П. Гладышев, Ю.А. Ершов, О. А.
Шустова. - М.: Химия, 1979. - 271 с.
Грасси,
Н. Деструкция и стабилизация полимеров / Н. Грасси. - М.: Мир, 1988. - 246 с.
Шляпников,
Ю.А. Антиокислительная стабилизация полимеров / Ю.А. Шляпников, С.Г. Кирюшкин,
А.П. Марьин. - М.: Химия, 1986. - 252 с.
Эмануэль,
Н.М. Цепные реакции окисления углеводородов в жидкой фазе / Н.М. Эмануэль, Е.Т.
Денисов, З.К. Майзус. - М.: Наука, 1965. - 367с.
Тугов,
И.И. Химия и физика полимеров / И.И. Тугов, Г.И. Кострыкина. - М.: Химия, 1989.
- 432 с.
Шилов,
Ю.Б. Кинетика и механизм инициированного окисления полипропилена и полиэтилена
в твердой фазе : диссертация канд. хим. наук. Черноголовка. 1979. - 210с.
Шилов,
Ю.Б. Высокомолекулярные соединения / Ю.Б. Шилов, Е.Т. Денисов. // Высокомол.
соед. - 1978. - Т. А.20. (№8). - С. 1849 - 1853.
Денисов,
Е.Т. // Высокомол. соед. - 1977. - Т. А19. (№11) - С. 2513 - 2523.
.
Лин, Д.Г. Исследование адгезии и окисления стабилизированного полиэтилена /
Д.Г. Лин // Известия АН БССР. Серия физ.-техн. наук. - 1978. - № 3. - С. 126.
Рейтлингер,
С.А. Проницаемость полимерных материалов / С.А. Рейтлингер. - М. : Химия, 1974.
- 174 с.
Денисов,
Е.Т. Реакции радикалов ингибиторов и механизм ингибированного окисления
углеводородов / Е. Т. Денисов. // Итоги науки и техники: сер. Кинетика и
катализ. - М.: ВИНИТИ, 1987. - Т. 17. С.3-115.
Керрингтон,
А. Магнитный резонанс и его применение в химии / А. Керрингтон, Э. Мак Лечлан.
- М.: Мир, 1970. - 447 с.
Слоним,
И.Я. Ядерный магнитный резонанс в полимерах / И.Я. Слоним, А.Н. Любимов. - М.:
Химия, 1966. - 337 с.
Ионин,
Б.И. ЯМР-спектроскопия в органической химии / Б.И. Ионин, Б.А. Ершов, А.И.
Кольцов. - 2-е изд. - Л.: Химия, 1983. - 272 с.
Козицына,
Л.А. Применение УФ- , ИК- и ЯМР-спектроскопии в органической химии / Л.А.
Козицына, Н.Б. Куплетская. - М.: Высшая школа, 1971. - 264 с.
Бураченко,
А.Л. Исследование полимеров методом парамагнитного зонда / А.Л. Бураченко, А.Л.
Коварский, А.М. Вассерман. // Успехи химии и физики полимеров. - М.: Химия,
1973. - 360 с.
Пшежецкий,
С.Я. ЭПР свободных радикалов в радиационной химии / С.Я. Пшежецкий, А.Г. Котов,
В.К. Милинчук и др. - М.: Химия, 1972. - 480 с.
.
Долгов, В.Н. Катализ в органической химии / В.Н. Долгов. - Л.: Госхимиздат,
1959. - 878 с.
Уайт,
Дж.Л. Полиэтилен, полипропилен и другие полиолефины / Дж.Л. Уайт, Д.Д. Чой /
пер. с англ. яз. под. ред. Е.С. Цобкалло - СПб.: Профессия, 2006. - 256 c.
Корягин,
С.И. Способы обработки материалов / С.И. Корягин, И.В. Пименов, В.К. Худяков. -
Учебное пособие / Калинингр. ун-т - Калининград, 2000. - 448 с.
.
Рогинский, В.А. Фенольные антиоксиданты: Реакционная способность и
эффективность / В.А. Рогинский - М.: Наука, 1988. - 247 с.
.
Шевцов, Г. А. Технология переработки пластических масс / Г. А. Шевцов, Д.У.
Алимова, М. Д. Барышникова - М.: Химия, 1988. - 512 с.
.
Кузьминский, А.С. Старение и стабидизация полимеров / А.С. Кузьминский - М.:
Химия, 1966. -212 с.
.
Горбунов, Б.Н. Химия и технология стабилизаторов полимерных материалов / Б.Н.
Горбунов, Я.А. Гурвич, И.П. Маслова - М.: Химия, 1981. - 368 с.
.
Ройтер, В.А. Cправочник: каталитические свойства веществ / В.А. Ройтер. - Киев:
АН УССР,1968. - 717 с
.
Ахметов, Н.С. Общая и неорганическая химия: учебник для вузов / Н.С. Ахметов -
М.: Высшая школа, 1998. - 368 с.
.
Вилков, Л.В. Физико-химические методы исследования в химии / Л.В. Вилков, Ю.А.
Пентин - М.: Высш. шк, 1987. - 367 с.
.
Тарутина, Л.И. Спектральный анализ полимеров / Л.И. Тарутина, Ф.О. Позднякова -
Л.: Химия, 1986. - 248 с.
.
Лин, Д.Г. Окисление ингибированного фенольным антиоксидантом полиэтилена в
условиях контакта с металлическим цинком / Д.Г. Лин, Е.В. Воробьева, Н.В.
Марченко // Журнал прикладной химии. - 2008. - Т. 81., Вып. 11. - С. 1866-1867.
.
Миндлин, С.С. Технология производства полимеров и пластических масс на их
основе / С. С. Миндлин - Л.: Химия, 1973. - 352 с.
.
Соколов, Р.С Химические технологии: учеб. пособие для студентов ВУЗ / Р.С.
Соколов - М.: Владос, 2000. - 448 с.
.
Гуль, В.С Физико-химические основы производства полимерных пленок / В.С Гуль,
В.П. Дьяконова - М.: Высшая школа, 1978. - 279 с.
.
Аскадский, А.А. Химическое строение и физические свойства полимеров / А.А.
Аскадский, Ю.И. Матвеев - М.: Химия,1983. - 248 с.
.
Лин, Д. Г. Контактное окисление ингибированного полиэтилена на меди при
неравномерном распределении антиоксиданта / Д. Г. Лин, Е. В. Воробьева //
Журнал прикладной химии. - 2011. - Т. 84, вып. 5. - С. 848-852.
.
Лин, Д.Г. Влияние антиоксидантов на окисление полиэтилена, катализируемое медью
/ Д.Г. Лин, Е.В. Воробьева // Химия и химическая технология. - 2005. - Т. 48. -
Вып. 12. - С. 61-66.
.
Каган, Д.Ф. Многослойные и комбинированные пленочные материалы / Д.Ф. Каган,
В.Е. Гуль, А.Д. Самарина - М.: Химия,1989.- 288 с.
.
Пинчук, П.С. Полимерные пленки, содержание, ингибиторы коррозии / П.С. Пинчук,
А.С. Неверов - М.: Химия, 1993. - 176 с.
41.
Глинка, Н.Л. Общая химия / Н.Л. Глинка - Л.: Химия, 1985. - 702 с.
42.
Лурье, Ю.Ю. Справочник по аналитической химии / Ю.Ю. Глинка - Л.: Химия, 1971.
- 453 с.
.
Береснева, В.С. Влияние валентности металла в составе оксида-добавки на
окисление полиэтилена, ингибированного ирганоксом 1010 // / В.С. Береснева //
Технология 2012: материалы Международной научно-технической конференции,
Северодонецк, 6-7 апреля 2012 г./ Технологический институт Восточноукраинского
национального университета им. В. Даля; ред. Тарасов В.Ю. - Северодонецк 2012.-
Ч.2.- С. 90-94.