|
Ключові слова
|
Ключове слово забезпечує
|
|
PM3
|
Розрахунок електронної структури буде
проведено цим методом
|
|
1SCF
|
Розрахунок електронної структури для заданої
геометричної конфігурації молекулярної системи в режимі статичної моделі
|
|
EF
|
Оптимізацію вказаних структурних елементів
хімічної частинки процедурою «Eigenvector Following»
за алгоритмом Бейкера
|
|
LET GNORM
|
Оптимізацію вказаних структурних елементів
хімічної частинки з вказаною граничною величиною норми градієнта, при
досягненні якої програма завершує процес розрахунку і переходить до видачі
результатів
|
|
CHARGE
|
Квантовохімічний розрахунок з вказаною
величиною заряду молекулярної системи або атома
|
|
DOUBLET
|
Розрахунок атомів, радикалів та іон-радикалів,
хімічних частинок з непарним числом електронів
|
|
T
|
Максимальний час в секундах, який відводиться
для квантовохімічного розрахунку
|
|
GRADIENTS
|
Виведення блоку інформації з величинами норми
градієнта для кожного структурного елемента, який оптимізувався
|
|
VECTORS
|
Виведення лише тієї частини блоку МО, яка
включає граничні МО
|
|
VECTORS ALLVEC
|
Виведення повного блоку МО
|
|
BONDS
|
Виведення блоку порядків зв’язків
|
|
ENPART
|
Розрахунок та виведення енергій двохатомних
взаємодій
|
|
FORCE THERMO(298) ISOTOPE ROT=1
|
Розрахунок та виведення ентропії
|
У роботі досліджувалися наступні
об'єкти:
- син та анти -
конформації пероксиазотистої кислоти;
- син та анти - конформації
дихлорметилпероксиазотистої кислоти.
Мета.
Освоєння методики моделювання заданої
конфігурації молекулярних систем.
Завдання до лабораторної роботи.
1.
Використовуючи програмний комплекс побудувати моделі син і анти-періпланарних
конформацій пероксиазотистої кислоти (O)NOOH
та дихлорметилпероксиазотистої кислоти (O)NOOCHCl2
відповідно до заданих молекулярних параметрів:
для sp-конформації
(O)NOOH:
<NOOH=90o,
<OON(O)=0o;
для ap-конформації
(O)NOOH:
<NOOH=90o,
<OON(O)=180o;
для sp-конформації
(O)NOOCHCl2:
<NOOC=90o,
<OON(O)=0o;
для ap-конформації
(O)NOOCHCl2:
<NOOC=90o,
<OON(O)=180o.
2.
Провести квантовохімічні розрахунки sp-
та ap- конформацій (O)NOOH
та (O)NOOCHCl2
в режимі оптимізації молекулярної геометрії, використовуючи програмний комплекс
MOPAC 97.
Результати виконаної роботи
Для того, щоб підготувати файл з
розширенням zmt
необхідно, використовуючи програму HyperChem,
побудувати 3D
модель молекулярного об’єкта. Отриману модель молекули зберегли у файлі з
розширенням zmt
( для цього в меню File
вибрали Save
As та розширення zmt).
Дала назву файлам син-(O)NOOH.zmt
та син-(O)NOOCHCl2.zmt,
антисин-(O)NOOH.zmt
та антисин-(O)NOOCHCl2.zmt.
У файлі з розширенням zmt
зберігається така інформація, як довжина хімічного зв’язку, величина валентного
кута, величина кута торсійного фрагмента.
Програма МОРАС працює з файлами, що
мають розширення dat.
Такий файл підготовлюється наступним чином: використовуючи програму Total
Commander, скопіювали
отриманий раніше zmt
файл завдяки функції «Копія (F5)»,
при цьому змінюючи розширення zmt
на dat.
У файлі з розширенням dat
зберігається така інформація: ключові слова, Z-матриця,
величини довжин зв’язку, валентних кутів, торсійних кутів.
Файл з розширенням dat
має ключові слова, які займають 2 рядка, інформацію користувача, яка займає 1
рядок, Z-матриця
починається з 4 рядка і закінчується 0 або пустою строчкою.
В ключових словах вказали:
MNDO - цим
методом розрахували електронну структуру;
EF
- оптимізацію молекулярної геометрії виконали процедурою Eigenvector
Following за
алгоритмом Бейкера;
LET
GNORM=0.01 - параметри
молекулярної геометрії розрахували з граничною величиною норми градієнта 0.01;
GRADIENTS
- вивели у файлі результатів величини норми
градієнта для кожного геометричного параметра, який оптимізується;
T=3600 - вказали необхідний час для
розрахунку в секундах;
VECTORS
ALLVEC - вказали,
щоб у файлі результатів виводився блок молекулярних орбіталей;
BONDS - вказали,
щоб у файлі результатів виводився блок порядків зв’язків;
ENPART
- вказали, щоб у файлі результатів виводився блок енергій двоатомних взаємодій,
розрахованих в наближенні Маллікена.
У третьому рядку вказали назву
речовини та її формулу.
Щоб організувати запуск на виконання
завдання в середовищі Total
Commander, а також
для проведення квантовохімічного розрахунку з використанням MOPAC:
курсором помітили файл МОРАС-97.еxe
і натиснули комбінацію клавіш Ctrl і Enter. В командному рядку з`явилось ім’я
файла МОРАС-97.exe.
Курсором помітили файл з розширенням dat
і натиснули комбінацію клавіш Ctrl і Enter. В командному рядку з`явилось ім’я
файла. Курсор перенесли в командний рядок і вилучили розширення dat
та крапку. Для запуску програми МОРАС-97 натиснули клавішу Enter. В робочому
вікні MS-DOS
з`явилася інформація про хід квантовохімічного розрахунку.
Короткий набір результатів
квантовохімічного розрахунку зберігається у файлі з розширенням arc.
Повний набір результатів
квантовохімічного розрахунку зберігається у файлі з розширенням out.
В arc
та out файлах значиться
така інформація - повна енергія, відстань між атомами, ентальпія, дипольні
моменти, енергія ВЗМО та НВМО та інше.
Висновки.
Виконавши лабораторну роботу я засвоїла методику
моделювання заданої конфігурації молекулярних систем на прикладі sp-
та ap-конформацій;
навчилась проводити квантовохімічний розрахунок молекул O)NOOH
та (O)NOOCHCl2;
навчилась працювати з програмами MOPAC
97 та Total
Commander;
ознайомилась зі структурою arc
та out файлів.
Лабораторна
робота № 2
Конформації
хімічної частинки і їх структурні параметри
Теоретична частина.
Існують такі функції, що описують
стан електрона, як атомна орбіталь, молекулярна орбіталь.
Атомна орбіталь - одноцентрова,
одноелектронна хвильова функція, що описує стан електрона в ефективному полі
ядра та інших електронів атома.
Молекулярна орбіталь -
багатоцентрова, одноелектронна хвильова функція, що описує стан електрона в
полі багатьох ядер та електронів молекулярної системи.
Базисні орбіталі - формально
визначені за допомогою спрощених рівнянь орбіталі, лінійна комбінація яких
дозволяє описати атомні та молекулярні орбіталі в лінійному наближенні.
Для розрахунку повної енергії
молекулярних систем використовують деякі наближення.
Повна енергія молекулярних систем у
наближенні Борна-Оппенгеймера - енергія молекулярної системи без урахування
кінетичної енергії руху ядер.
МО ЛКАО - наближення, в якому кожна
молекулярна орбіталь розглядається у вигляді лінійної комбінації атомних
орбіталей.
Валентне наближення розрахунку
електронної структури - наближення, в якому враховуються лише валентні
електрони.
Електронна енергія молекулярних
систем - сума потенціальних енергій взаємодій: електрон-електрон,
електрон-ядро; та кінетичних енергій руху кожного електрона.
Стандартна ентальпія утворення
молекулярних систем - термодинамічна характеристика, що являє собою тепловий
ефект реакції утворення даної сполуки з простих речовин.
Ядерно-електронний «остов» -
поняття, яке використовується в квантовохімічних розрахунках валентного
наближення. У такому наближенні допускається, що атомне ядро та невалентні електрони
атома утворюють атомний (ядерно-електронний) остов, заряд визначається
сумуванням заряду атомного ядра та заряду всіх невалентних електронів.
Валентні електрони - електрони,
здатні до утворення хімічних зв'язків.
Граничні МО - найвища зайнята молекулярна
орбіталь та найнижча вільна молекулярна орбіталь.
НЗМО - молекулярна орбіталь, що має
найвищу енергію з усіх зайнятих електронами МО.
НВМО - молекулярна орбіталь, що має
найнижчу енергію з усіх незаселених електронами молекулярних орбіталей хімічної
частинки.
Потенціал іонізації (за теоремою
Купманса) - це енергія НЗМО, взята с протилежним знаком.
Спорідненість до електрону -
властивість молекул приєднувати електрон; визначається енергією, що виділяється
при приєднанні електрона до молекули з утворенням аніон-радикалу.
Електронна густина - число
електронів в одиниці об'єму .
Електричний заряд на атомі -
розраховується за виразом: qA
=
ZA - QA,
де: ZA
- заряд ядра атома А, QA
- електронна густина на атомі А.
Довжина хімічного зв'язку - відстань
у рівноважному стані між положеннями ядер атомів, сполучених хімічним зв'язком.
Торсійний кут - стереохімічний
елемент молекулярної структури (двогранний кут).
Електронна заселеність (АО) -
вирахуване число електронів на певній атомній орбіталі, яка центрована на атомі
А.
Дипольний момент - вектор, що
характеризує зарядову асиметрію і умовно спрямований від центра негативного до
центра позитивного заряду.
Варіаційні коефіцієнти -
коефіцієнти, що показують внесок атомної орбіталі в загальну хвильову функцію.
Вікове рівняння - рівняння виду:

Мета.
Освоєння методики визначення стереохімічних параметрів та електронної будови
молекулярних систем за результатами квантовохімічних розрахунків (метод MNDO).
Завдання до роботи.
1. Дати
структурно хімічну характеристику молекули.
2. Освоїти
організацію структури файлу вхідних даних для квантовохімічного розрахунку.
3. Освоїти
організацію структури архівного (arc)
файлу результатів квантовохімічного розрахунку.
4. Освоїти
організацію структури архівного (оut)
файлу результатів квантовохімічного розрахунку.
5. Параметри
молекулярної геометрії та електронної структури
конформерів занести в таблицю.
6. Провести
аналіз впливу конформації пероксикислот на параметри їх молекулярної геометрії
та електронної структури.
Результати виконаної роботи
Таблиця 2.1.
Структурно-хімічна характеристика хімічник частинок
|
Параметри
|
(O)NOOH
|
(O)NOOCHCl2
|
|
кількість атомів
|
N
|
5
|
8
|
|
Кількість параметрів молекулярної геометрії
|
3N-6
|
9
|
18
|
|
кількість валентних зв`язків
|
N-1
|
4
|
7
|
|
кількість валентних кутів
|
N-2
|
3
|
6
|
|
кількість торсійних кутів
|
N-3
|
2
|
5
|
|
кількість зв`язуючих
електронних пар
|
nA
|
5
|
8
|
|
кількість незв`язуючих
електронних пар
|
nB
|
7
|
13
|
|
кількість АО у валентному наближенні
|
|
17
|
29
|
|
кількість електронів у валентному наближенні
|
|
24
|
42
|
|
кількість МО
|
2nA+nB
|
17
|
29
|
|
кількість зайнятих електронами МО
|
nA+nB
|
12
|
21
|
|
кількість вакантних МО
|
nA
|
5
|
8
|
|
Граничні МО
|
|
12 і 13
|
21 і 22
|
|
ВЗМО
|
|
12
|
21
|
|
НВМО
|
|
13
|
22
|
|
Підграничні МО
|
|
12 і 15
|
22 і 25
|
|
Спінова мультиплетність
|
2S+1
|
1
|
1
|
Таблиця 2.2.
Параметри молекулярної геометрії та електронної густини конформерів
пероксикислот
|
Параметри
|
(O)NOOH
|
(O)NOOCHCl2
|
|
|
Конформація ООN(О)
молекулярного фрагменту
|
Конформація ООN(О)
молекулярного фрагменту
|
|
sp
|
ap
|
sp
|
ap
|
|
rO-O,Å
|
1.30
|
1.29
|
1.30
|
1.29
|
|
rC-O,Å
|
1.32
|
1.34
|
1.32
|
1.34
|
|
rO-H,Å
|
0.96
|
0.96
|
1.40
|
1.41
|
|
<COOH,
град
|
90
|
90
|
90
|
90
|
|
∆Hf0,
ккал/моль
|
-21.26
|
-20.31
|
-27.15
|
-25.81
|
|
Ee, eB
|
-2838.94
|
-2811.42
|
-6118.88
|
-6075.66
|
|
EN,N , eB
|
1655.04
|
1627.55
|
4098.05
|
4054.89
|
|
Etot , eB
|
-1183.90
|
-1183.87
|
-2020.83
|
-2020.77
|
|
μ,
D
|
1.81
|
1.92
|
1.80
|
1.31
|
|
EНЗМО,
еВ
|
-11.85
|
-11.58
|
-12.35
|
-12.09
|
|
EНВМО, еВ
|
-0.32
|
-0.38
|
-0.82
|
-0.82
|
|
pO-O
|
1.29
|
1.29
|
1.48
|
1.48
|
|
qO1, e
|
-0.13
|
-0.14
|
-0.14
|
-0.14
|
|
qO2, e
|
-0.18
|
-0.13
|
-0.15
|
-0.11
|
Повна енергія хімічної частини
розраховується по формулі:
Etot
= Ee
+ EN,N
Розрахуємо повну енергію для sp-конформації
та ap-конформації
молекули (O)NOOH:
Etot
(sp)
= -2838.94+
1655.04= -1183.90
еВ
Etot
(ap) = -2811.42+
-4055.00= -1183.87
еВ
Розрахуємо повну енергію для sp-конформації
та ap-конформації
молекули (O)NOOCHCl2H:
Etot
(sp)
= -6118.88 + 4098.05= -2020.83 еВ
Etot
(ap)
= -6075.66 + 4054.89= -2020.77
еВ
Висновки.
Виконавши лабораторну роботу я освоїла
методику визначення параметрів стереохімічної та електронної будови
молекулярних систем за результатами квантовохімічних розрахунків молекул (O)NOOH
та (O)NOOCHCl2.
Аналізуючи данні, узяті з arc
та out файлів, які
приведені в таблиці 2, можна зробити висновок, що вплив конформації на
параметри молекулярної геометрії та електронної структури пероксикислот найбільш
вбачається в значеннях ентальпії, загальної енергії та її складових, дипольного
моменту, зарядів на атомах кисню. Проте, вид конформації пероксикислот не
впливає на значення міжатомних відстаней, торсійних кутів, вони при всіх
конформаціях залишаються майже незмінними величинами.
Лабораторна
робота № 3
Квантовохімічний
розрахунок в режимі координати внутрішнього обертання
Теоретична частина.
Часто речовина складається з суміші
конформерів - найбільш стійких конформацій.
Конформація - одне з неідентичних
розташувань у просторі груп атомів даної молекулярної частинки, що утворюється
під час обертання навколо одинарного зв'язку без його розриву зі збереженням
стереохімічної конфігурації молекули.
Бар'єр внутрішнього обертання -
різниця між величиною повної енергії молекулярної конформації в максимумі на
шляху обертання атома чи групи атомів навколо хімічного зв'язку та повною
енергією конформера.
Бар'єр внутрішнього обертання
визначається за формулою:
∆VA-B Etot
(C) - Etot (A)
∆VB-A Etot
(C) - Etot (B)
Для вивчення конформацій молекули та
виявлення її конформерів у програмі МОРАС використовується режим координати
внутрішнього обертання - режим квантовохімічного розрахунку, у якому поступово
змінюється величина будь-якого торсійного кута молекулярного фрагмента.
Мета.
Освоєння методики квантовохімічних розрахунків молекулярних систем у режимі
координати внутрішнього обертання.
Завдання до роботи.
Провести квантовохімічний розрахунок
у режимі координати внутрішнього обертання ряду конформацій вивчаємих молекул,
змінюючи величину торсійного кута -О-О-N=О
молекулярного фрагменту в діапазоні від 0 до 360 градусів з кроком 30 градусів.
Значення стандартних ентальпій утворення, дипольних моментів та торсійного кута
NООН для отриманої
послідовності конформацій занести в таблицю.
Результати виконаної роботи
Файли син-(O)NOOH.dat
та син-(O)NOOCHCl2.dat
перейменували на Динаміка_син-(O)NOOH.dat
та Динаміка_син-(O)NOOCHCl2.dat.
У файлах (O)NOOH.arc
та (O)NOOCHCl2.arc,
де сберігається структурна інформація син-рівноважної конфігурації
пероксикислоти, виділили і скопіювали в буфер памяті блок інформації, починаючи
з рядка ключових слів і завершуючи Z-матрицею.
У файлах Динаміка_син-(O)NOOH.dat
та Динаміка_син-(O)NOOCHCl2.dat
виділили всю інформацію і вставили блок інформації із буфера памяті. Отримані
файли Динаміка_син-(O)NOOH.dat
та Динаміка_син-(O)NOOCHCl2.dat
мають завершуватись нулем.
В Z-матриці
вибрали структурний елемент, величину якого потрібно послідовно змінювати -
торсійний кут для О4, тобто <ООN(О).
Для нього вказали мітку координати внутрішнього обертання (-1).
Також вказали послідовність значень
координати внутрішнього обертання: 0, 30, 60, 90, 120, 150, 180, 210, 240, 270,
300, 330, 360 градусів. Значення відокремлили одним пробілом.
Виконали квантовохімічний розрахунок
за допомогою MOPAC 97 та
виявили конформери сполук. Усі дані занесли в таблицю 3.1.
Таблиця 3.1. Стандартні
ентальпії утворення, дипольні моменти і величини кутів NООН
і NOOC.
|
<ООN(О),
град
|
(O)NOOH
|
(O)NOOCHCl2
|
|
|
∆Hf0
ккал/моль
|
μ, D
|
<NOOH,
град
|
∆Hf0
ккал/моль
|
μ, D
|
<NOOC,
град
|
|
0
|
-21.25
|
1.82
|
147.26
|
-27.19
|
1.86
|
147.26
|
|
30
|
-19.65
|
1.61
|
227.45
|
-25.20
|
1.74
|
144.25
|
|
60
|
-15.53
|
1.40
|
229.28
|
-21.35
|
1.55
|
128.96
|
|
90
|
-12.64
|
1.33
|
226.81
|
-29.56
|
1.57
|
118.49
|
|
120
|
-13.89
|
1.34
|
218.98
|
-21.35
|
1.58
|
116.25
|
|
150
|
-17.77
|
1.61
|
219.64
|
-24.56
|
1.41
|
119.43
|
|
180
|
1.89
|
239.40
|
-25.76
|
1.32
|
129.37
|
|
210
|
-19.07
|
1.95
|
249.39
|
-23.49
|
1.35
|
137.36
|
|
240
|
-15.56
|
1.77
|
251.39
|
-19.85
|
1.47
|
144.64
|
|
270
|
-13.42
|
1.59
|
246.05
|
-18.62
|
1.49
|
147.80
|
|
300
|
-15.18
|
1.59
|
230.67
|
-21.33
|
1.56
|
149.21
|
|
330
|
-19.65
|
1.61
|
227.43
|
-25.48
|
1.77
|
148.24
|
|
360
|
-21.25
|
1.82
|
147.26
|
-27.19
|
1.91
|
147.26
|
Висновки.
Виконавши лабораторну роботу я засвоїла методику
квантовохімічних розрахунків молекул(O)NOOH
та (O)NOOCHCl2
у режимі координати внутрішнього обертання. Отримані результати брала из arc
та out
файлів. Отримані значення занесла в таблицю. Згідно таблиці можна зробити
висновок, що конформерами як у пероксиазатистої кислоти, так і у
дихлорметилпероксиазотистої кислоти, є конфігурації з торсійним
кутом ОNО(О)
0 та 180 градусів.
Лабораторна
робота № 4
Ентропія
хімічних сполук
Теоретична частина.
Третій закон термодинаміки - поблизу
абсолютного 0 усі термодинамічні процеси протікають без зміни ентропії.
Для того, щоб розрахувати значення
вільної енергії Гіббса необхідно скористатися наступним рівнянням: ∆G
= ∆H - T∆S.
Ентропія -
Мета.
Освоєння методики розрахунку ентропії хімічних сполук.
Завдання до роботи.
1. Розрахувати
абсолютну ентропію конформерів пероксикислот.
2. Значення
величин абсолютної ентропії і її компонентів занести в таблицю.
3. Розрахувати
вільну енергію Гіббса для конформерів при 298К, результати занести в таблицю.
Результати виконаної роботи
Абсолютну ентропію можна розрахувати
тільки для рівноважного стану хімічних сполук.
Щоб розрахувати абсолютну ентропію
ми провели квантовохімічний розрахунок об’єктів з оптимізацією молекулярної
геометрії. Z-матрицю
рівноважного стану об’єктів скопіювали в архівних файлах arc
в буфер пам’яті та замінили Z-матрицю
початкових даних у файлах з розширенням dat.
Отримали файли даних та відреагували
необхідний набор ключових слів для розрахунку ентропії: AM1
T=3600 FORCE
THERMO (298) isotope
rot=1.
Даним файлам дали назву Ентропія_син-(O)NOOH.dat
та Ентропія_син-(O)NOOCHCl2.dat.
Файли з розширенням dat
і out скопіювали у свою
папку.
Результати розрахунку знаходяться в out
файлі.
Отримані значення ентропії занесли в
таблиці 4.1. та 4.2.
Таблиця 4.1.
Абсолютна ентропія конформерів (O)NOOH
та (O)NOOCHCl2
та їх складові.
|
Ентропія, кал/(кмоль)
|
(O)NOOH
|
(O)NOOCHCl2
|
|
|
Конформер А
|
Конформер Б
|
Конформер А
|
Конформер Б
|
|
Sкол298
|
4.68
|
4.84
|
8.60
|
9.29
|
|
Sоб298
|
23.08
|
22.66
|
29.00
|
29.12
|
|
Sпост298
|
38.34
|
38.34
|
40.84
|
40.84
|
|
S298
|
66.10
|
65.84
|
78.44
|
79.25
|
молекулярний хімічний
частина конформація
Таблиця 4.2.
Термодинамічні параметри конформерів (O)NOOH
та (O)NOOCHCl2
|
Термодинамічні параметри
|
(O)NOOH
|
(O)NOOCHCl2
|
|
|
Конформер А
|
Конформер Б
|
Конформер А
|
Конформер Б
|
|
∆Hf0,
ккал/моль
|
-12.06
|
-6.34
|
-12.25
|
-7.07
|
|
S298,
кал/(кмоль)
|
66.10
|
65.84
|
78.44
|
79.25
|
|
∆G,
ккал/моль
|
-31.75
|
-25.96
|
-35.63
|
-30.69
|
Розрахуємо значення вільної енергії
Гіббса за формулою:
∆G
= ∆H
- T∆S
Розрахуємо вільну енергію Гіббса для
конформерів А и Б молекули (O)NOOH:
∆G
= -12.06- 298*66.10*103=
-31.75, (ккал/моль)
 ∆G = -6.34-
298*65.84*103= -25.96, (ккал/моль)
∆G = -6.34-
298*65.84*103= -25.96, (ккал/моль)
Розрахуємо вільну енергію
Гіббса для конформерів А и Б молекули (O)NOOCHCl2:
∆G = -12.25- 298*78.44*103= -35.63, (ккал/моль)
-12.25- 298*78.44*103= -35.63, (ккал/моль)
∆G = -7.07- 298*79.25*103
= -30.69, (ккал/моль)

Висновки. Виконавши
цю роботу, мною була засвоєна методика розрахунку ентропії хімічних сполук.
Також були знайдені ентропії досліджуємих пероксикислот (O)NOOH та
(O)NOOCHCl2.
Розрахувала вільну енергію Гіббса конформерів А і Б досліджуємих молекул.
Аналізуючи данні,які наведені в таблиці 4.2., можна зробити висновок, що
значення вільної енергії Гіббса майже не залежить від виду конформації
пероксикислоти.
Лабораторна
робота №5
Конформаційний
аналіз хімічних сполук
Теоретична частина.
Хімічні та біологічні функції
хімічних частинок речовини пов'язані з їх просторовою та електронною будовою.
Саме тому великого значення набуває конформаційний аналіз.
Конформаційний аналіз - визначення
фізичних і хімічних властивостей конформерів хімічних частинок речовини в
основному, перехідному, а також у збудженому станах; встановлення зв'язку між
реакційною здатністю чи властивостями конформерів хімічних частинок речовини з
параметрами їх електронної будови.
Часто речовина складається з суміші
конформерів - найбільш стійких конформацій, що знаходяться у рівновазі між
собою.
Конформація - одне з неідентичних
розташувань у просторі груп атомів даної молекулярної частинки, що утворюється
під час обертання навколо одинарного зв'язку без його розриву зі збереженням
стереохімічної конфігурації молекули.
Конформери - конформаційні ізомери,
яким відповідають мінімуми на поверхні потенційної енергії, і які знаходяться у
рівноважному стані, хоча не можуть бути виділені через невисокий енергетичний
бар'єр обертання.
Конформаційна енергія - різниця
повних енергій довільної конформації та конформера з найнижчою енергією.
Один конформер може перейти в інший,
або навпаки, при цьому йому потрібно здолати бар'єр внутрішнього обертання.
Бар'єр внутрішнього обертання -
різниця між величиною повної енергії молекулярної конформації в максимумі на
шляху обертання атома чи групи атомів навколо хімічного зв'язку та повною
енергією конформера.
В залежності від значення торсійного
кута молекулярного фрагменту існує конформаційна термінологія.
Конформаційна термінологя: Θ
= 0 градусів - цис-конформація;
Θ = 180 градусів
- анти-конформація;
Θ ≠ 0 або
180 градусів - гош-коформація;
Θ ≠ 0 або
180 градусів - S-конформація.
Мета.
Освоєння методики конформаційного аналізу.
Завдання до роботи.
Провести конформаційний аналіз
пероксикислот (O)NOOH
та (O)NOOCHCl2.
Результати виконаної роботи
Значення ентальпії ∆Hf0,
ентропії S298
та вільної енергії Гіббса взяли з таблиці 4.1.
попередньої лабораторної роботи.
Побудували график залежності
стандартної єнтальпії утворення конформацій (O)NOOH
та (O)NOOCHCl2
від величини торсійних кутів OON(O)
фрагмента, використовуючи дані з таблиці 3.1.
Рис. 5.1. Зміна
стандартної ентальпії утворення (O)NOOH
вздовж координати внутрішнього обертання ацильного фрагмента довкола N-O
зв’язку.

Рис. 5.2. Зміна
стандартної ентальпії утворення (O)NOOCHCl2
вздовж координати внутрішнього обертання
ацильного фрагмента довкола N-O
зв’язку.

Рис.5.3.
Залежність кута NООН
від координати внутрішнього обертання ацильного фрагменту довкола звязку N-O
для молекули (O)NOOH.

Рис.5.4. Залежність
кута NООC
від координати внутрішнього обертання для ацильного фрагменту довкола зв’язку N-O
для молекули (O)NOOCHCl2.

Визначила число конформерів - 2.
Розрахувала величину барьєрів внутрішнього обертання ацильного фрагменту за
формулами:
∆VA-B Etot
(C) - Etot (A)
∆VB-A Etot
(C) - Etot (B)
Для пероксиазотистої кислоти (O)NOOH:
∆VA-B
=-12.64
- (-20.25)
= 7.61
ккал/моль
∆VB-A
= -13.42
- (-20.25)
= 6.83
ккал/моль
Для дихлорпероксиазотистої кислоти (O)NOOCHCl2:
∆VA-B
= -19.56
- (-25.76)
= 6.2
ккал/моль
∆VB-A
= -18.62
- (-25.76)
= 7.14
ккал/моль
Константа конформаційної рівноваги
для конформерів (O)NOOH
та (O)NOOCHCl2
розраховується за формулою:

Або: Кр = exp( )
)
Щоб розрахувати константу,
потрібно знати значення ∆H та ∆S
для
молекул (O)NOOH та
(O)NOOCHCl2.
Розрахуємо значення ∆H для
молекул (O)NOOH та
(O)NOOCHCl2:
∆H (O)NOOH =
-6.34 -
(-12.06) = 5.72 ккал/моль
= 5.72*103 кал/моль
∆H(O)NOOСHCl2 =
-7.07 - (-12.25) = 5.18 ккал/моль = 5,18*103 кал/моль
Розрахуємо значення ∆S для
молекул (O)NOOH та
(O)NOOCHCl2:
∆S (O)NOOH =
65,84 - 66,10 = -0,26 кал/моль
∆S (O)NOOСHCl2 =
79,25 - 78,44 = 0,81 кал/моль
Розрахуємо константу конформаційної
рівноваги для молекули (O)NOOH:
Kp = exp ( ) = 7.04*10-5
) = 7.04*10-5
Розрахуємо константу
конформаційної рівноваги для молекули (O)NOOCHCl2:
Kp = exp ( ) = 1.03*10-5
) = 1.03*10-5
Розрахувала кількісний склад
рівноважної конформаційної суміші як (O)NOOH так
і (O)NOOCHCl2.
Склад конформерів А і Б для (O)NOOH:
nA =  .
.
Тоді склад конформера Б: nB = 0.
Склад конформерів А і Б для (O)NOOCHCl2:
nA = =100%.
=100%.
Тоді склад конформера Б: nB = 0.
Таблиця 5.5.
Результати конформаційного аналізу молекул (O)NOOH
та (O)NOOCHCl2
|
Параметри
|
(O)NOOH
|
(O)NOOCHCl2
|
|
|
А
|
Б
|
А
|
Б
|
|
Число конформерів
|
2
|
2
|
|
∆Hf0,
ккал/моль
|
-12,06
|
-6.34
|
-12.25
|
-7.07
|
|
S298,
кал/(кмоль)
|
66,10
|
65.84
|
78.44
|
79.25
|
|
∆G,
ккал/моль
|
-31,75
|
-25.96
|
-35.63
|
-30.69
|
|
∆VA-B,
ккал/моль
|
7.61
|
6.2
|
|
∆VВ-А,
ккал/моль
|
6.83
|
7.14
|
|
Кр
|
7,04*10-5
|
1,03*10-5
|
|
Вміст конформерів, %
|
100
|
0
|
100
|
0
|
Висновки.
Виконавши цю лабораторну роботу, я засвоїла методику конфірмаційного аналізу та
навчилась розраховувати константу конфірмаційної рівноваги для молекул (O)NOOH
та (O)NOOCHCl2,
навчилась розраховувати бар’єри внутрішнього обертання. Побудувала графіки
залежності координати внутрішнього обертання від стандартної ентальпії
утворення та від кута NOOH
та NOOC.
Лабораторна
робота № 6
Розподіл
електронної густини в хімічних сполуках
Теоретична частина.
Граничні МО - найвища зайнята
молекулярна орбіталь та найнижча вільна молекулярна орбіталь.
НЗМО - молекулярна орбіталь, що має
найвищу енергію з усіх зайнятих електронами МО.
НВМО - молекулярна орбіталь, що має
найнижчу енергію з усіх незаселених електронами молекулярних орбіталей хімічної
частинки.
Потенціал іонізації (за теоремою
Купманса) - це енергія НЗМО, взята с протилежним знаком.
Спорідненість до електрону -
властивість молекул приєднувати електрон; визначається енергією, що виділяється
при приєднанні електрона до молекули з утворенням аніон-радикалу.
Електронна густина - число
електронів в одиниці об'єму .
Електричний заряд на атомі -
розраховується за виразом: qA
=
ZA - QA,
де: ZA
- заряд ядра атома А, QA
- електронна густина на атомі А.
Довжина хімічного зв'язку - відстань
у рівноважному стані між положеннями ядер атомів, сполучених хімічним зв'язком.
Торсійний кут - стереохімічний
елемент молекулярної структури (двогранний кут).
Електронна заселеність (АО) -
вирахуване число електронів на певній атомній орбіталі, яка центрована на атомі
А.
Дипольний момент - вектор, що
характеризує зарядову асиметрію і умовно спрямований від центра негативного до
центра позитивного заряду.
Варіаційні коефіцієнти -
коефіцієнти, що показують внесок атомної орбіталі в загальну хвильову функцію.
Мета.
Освоєння методики розрахунку параметрів електронної структури: електронної
заселеності АО, електронної густини та електричного заряду на атомах хімічної
частини у наближенні Маллікена.
Завдання до роботи.
1. Розрахувати
електронну заселеність s,
px, py
та pz АО атомів О1
і Н5 вивчаємої молекули (sp
- рівноважна молекулярна конформація).
2. Розрахувати
електронну густину на атомах O1і
Н5 вивчаємої молекули (sp
- рівноважна молекулярна конформація).
3. Розрахувати
електричний заряд на атомах O1і
Н5
вивчаємої
молекули (sp
- рівноважна молекулярна конформація).
Блок молекулярних орбіталей м-окси-п-метилпероксибензойної
кислоти.
Root No. 1 2 3 4 5 6 7 8
A 2 A 3 A 4 A 5 A 6 A 7 A 8 A
.542 -38.750 -36.567 -35.226 -33.807
-32.655 -28.995 -26.930O 1 -0.0317 -0.1719 0.1747 -0.5990 -0.5685 0.2700 0.0086
0.0660O 1 -0.0153 -0.0582 0.0323 -0.0746 0.0485 -0.0550 -0.0067 -0.0741O 1
-0.0060 -0.0172 0.0064 0.0035 0.0118 -0.0103 0.0009 0.0046O 1 -0.0054 -0.0269
0.0249 -0.0767 -0.0679 0.0295 -0.0017 0.0040H 5 -0.0237 -0.1020 0.0853 -0.2624
-0.2140 0.0921 0.0009 0.0150
Root No. 9 10 11 12 13 14 15 16
A 10 A 11 A 12 A 13 A 14 A 15 A 16 A
.539 -22.002 -21.058 -19.025 -18.365
-18.182 -17.377 -16.617O 1 -0.0455 0.0057 0.0334 0.0096 0.0134 0.0661 0.2321
0.0420O 1 0.1153 -0.0369 -0.1169 0.0551 -0.2852 0.3323 -0.0949 0.1045O 1
-0.0418 0.0157 0.0680 -0.1503 0.0134 0.0632 0.0456 0.1262O 1 -0.0043 0.0151
0.0054 -0.0375 0.1214 -0.3206 -0.4991 -0.1405H 5 -0.0103 0.0098 0.0086 -0.0172
0.1032 -0.2316 -0.2967 -0.1023
Root No. 17 18 19 20 21 22 23 24
A 18 A 19 A 20 A 21 A 22 A 23 A 24 A
.511 -15.595 -15.413 -15.047 -14.966
-14.821 -14.246 -13.995O 1 0.1062 0.0314 0.0990 -0.0783 0.0167 0.1147 -0.0105
-0.0108O 1 -0.1432 0.0160 -0.1715 0.1337 0.3294 -0.1464 -0.0105 -0.0606O 1
-0.0534 -0.0266 -0.0367 0.0360 0.0665 -0.0723 -0.0235 0.0259O 1 -0.2019 -0.0869
-0.2073 0.1743 -0.1208 -0.2735 0.0279 0.0359H 5 -0.1098 -0.0551 -0.1124 0.0949
-0.1151 -0.1560 0.0208 0.0314
Root No. 25 26 27 28 29 30 31 32
A 26 A 27 A 28 A 29 A 30 A 31 A 32 A
.440 -12.987 -12.960 -12.688 -12.200
-11.402 -10.144 -9.488O 1 -0.0080 0.0147 -0.0362 0.0152 0.0661 0.0012 -0.0020
-0.0006O 1 0.0748 -0.0551 0.0829 0.0328 -0.0112 0.0203 0.0168 0.0149O 1 0.0487
0.6781 -0.4257 -0.4810 -0.1084 0.1976 0.0235 0.0093O 1 0.0254 0.0139 0.0629
-0.0737 -0.1829 -0.0384 0.0092 0.0041H 5 0.0010 -0.0190 0.0541 -0.0335 -0.1298
-0.0084 0.0025 0.0006
Root No. 33 34 35 36 37 38 39 40
A 34 A 35 A 36 A 37 A 38 A 39 A 40 A
.551 0.026 0.481 1.052 2.150 2.281
2.404 2.780O 1 0.0192 -0.0014 0.0973 0.0209 -0.0727 0.0148 -0.0411 -0.0036O 1
0.1603 -0.0067 0.5934 0.0911 -0.2357 0.0542 -0.1165 -0.0060O 1 0.0177 0.0004
0.0379 0.0081 0.0120 -0.0042 0.0101 0.0021O 1 0.0355 -0.0008 0.1285 0.0111
-0.1024 0.0224 -0.0618 -0.0071H 5 -0.0104 0.0015 -0.0875 -0.0317 0.1038 -0.0187
0.0619 0.0057
Root No. 41 42 43 44 45 46 47 48
A 42 A 43 A 44 A 45 A 46 A 47 A 48 A
.058 3.133 3.323 3.692 3.841 3.882
3.996 4.198O 1 -0.0392 -0.0015 0.0182 0.0111 0.0216 -0.0039 -0.0439 -0.1663O 1
-0.0692 -0.0061 0.0265 0.0067 0.0117 -0.0022 -0.0056 0.0669O 1 0.0108 0.0009 -0.0057
-0.0046 -0.0064 0.0014 0.0140 0.0396O 1 -0.0640 -0.0013 0.0306 0.0213 0.0424
-0.0079 -0.0922 -0.3856H 5 0.0750 0.0021 -0.0382 -0.0268 -0.0552 0.0101 0.1218
0.5201
Root No. 49 50 51 52 53 54 55 56
A 50 A 51 A 52 A 53 A 54 A 55 A 56 A
.463 4.539 4.623 4.862 5.148 5.706
5.881 6.869O 1 -0.1283 0.0114 -0.0353 -0.0231 -0.0068 -0.0002 -0.0004 0.0073O 1
0.1692 -0.0217 0.0637 0.0813 0.0508 0.0111 -0.0145 0.0729O 1 0.0130 -0.0010
-0.0015 -0.0085 -0.0071 -0.0031 0.0021 -0.0155O 1 -0.3489 0.0330 -0.1067 -0.0879
-0.0389 -0.0071 0.0041 -0.0084 H
5 0.4830 -0.0456 0.1508 0.1266 0.0573 0.0112 -0.0067 0.0190
ρ(О1)S
=
(-0.0317)2 + (-0.1719)2 + (0.1747)2 +
(-0.5990)2 + (-0.5685)2 + (0.2700)2+ +
(0.0086)2 + (0.0660)2 + (-0.0455)2 + (0.0057)2
+ (0.0334)2 + (0.0096)2 + + (0.0134)2 +
(0.0661)2 + (0.2321)2 + (0.0420)2 + (0.1062)2
+ (0.0314)2 + + (0.0990)2 + (-0.0783)2 +
(0.0167)2 + (0.1147)2 + (-0.0105)2 + (-0.0108)2
+ + (-0.0080)2 + (0.0147)2 + (-0.0362)2
+ (0.0152)2 + (0.0661)2 + (0.0012)2 + +
(-0.0020)2 + (-0.0006)2 = 1.8640
ρ(О1)Px
= (-0.0153)2 + (-0.0582)2 + (0.0323)2 +
(-0.0746)2 + (0.0485)2 + + (-0.0550)2 +
(-0.0067)2 + (-0.0741)2 + (0.1153)2 +
(-0.0369)2 + (-0.1169)2 + + (0.0551)2 +
(-0.2852)2 + (0.3323)2 + (-0.0949)2 + (0.1045)2
+ (-0.1432)2 + + (0.0160)2 + (-0.1715)2 +
(0.1337)2 + (0.3294)2 + (-0.1464)2 + (-0.0105)2
+ + (-0.0606)2 + (0.0748)2 + (-0.0551)2 +
(0.0829)2 + (0.0328)2 + (-0.0112)2 + +
(0.0203)2 + (0.0168)2 + (0.0149)2 = 0.9672
ρ(О1)Pу
= (-0.0060)2 + (-0.0172)2 + (0.0064)2 +
(0.0035)2 + (0.0118)2 + + (-0.0103)2 +
(0.0009)2 + (0.0046)2 + (-0.0418)2 + (0.0157)2
+ (0.0680)2 + + (-0.1503)2 + (0.0134)2 +
(0.0632)2 + (0.0456)2 + (0.1262)2 + (-0.0534)2
+ + (-0.0266)2 + (-0.0367)2 + (0.0360)2 +
(0.0665)2 + (-0.0723)2 + (-0.0235)2 + +
(0.0259)2 + (0.0487)2 + (0.6781)2 + (-0.4257)2
+ (-0.4810)2 + (-0.1084)2 + + (0.1976)2 +
(0.0235)2 + (0.0093)2 = 1.9908
ρ(О1)Pz
= (-0.0054)2 + (-0.0269)2 + (0.0249)2 +
(-0.0767)2 + (-0.0679)2 + + (0.0295)2 +
(-0.0017)2 + (0.0040)2 + (-0.0043)2 + (0.0151)2
+ (0.0054)2 + + (-0.0375)2 + (0.1214)2 +
(-0.3206)2 + (-0.4991)2 + (-0.1405)2 +
(-0.2019)2 + + (-0.0869)2 + (-0.2073)2 +
(0.1743)2 + (-0.1208)2 + (-0.2735)2 + (0.0279)2
+ + (0.0359)2 + (0.0254)2 + (0.0139)2 +
(0.0629)2 + (-0.0737)2 + (-0.1829)2 + +
(-0.0384)2 + (0.0092)2 + (0.0041)2 = 1.3183
ρ(H5)S
= (-0.0237)2
+ (-0.1020)2 + (0.0853)2 + (-0.2624)2 +
(-0.2140)2 + + (0.0921)2 + (0.0009)2 +
(0.0150)2 + (-0.0103)2 + (0.0098)2 + (0.0086)2
+ + (-0.0172)2 + (0.1032)2 + (-0.2316)2 +
(-0.2967)2 + (-0.1023)2 + (-0.1098)2 + +
(-0.0551)2 + (-0.1124)2 + (0.0949)2 +
(-0.1151)2 + (-0.1560)2 + (0.0208)2 + +
(0.0314)2 + (0.0010)2 + (-0.0190)2 + (0.0541)2
+ (-0.0335)2 + (-0.1298)2 + + (-0.0084)2 +
(0.0025)2 + (0.0006)2 = 0.8040
= Z - Q
QO1
=1.8640 + 0.9672
+ 1.9908 + 1.3183
= 6.1403
qO1
= 6 - 6.1403 = -0.1403
QН5
= 0.8040
qН5
= 1 - 0.8040 = 0.196
Висновки. Виконавши
цю роботу, мною були розраховані параметри електронної структури п-метил-м-оксипероксибензойної
кислоти: електронна заселеність АО, електронна густина та електричний заряд на
атомах О1 та Н5 пероксикислоти у наближенні Маллікена.
Також була визначена зв'язуюча МО, що контролює зв'язок О1-Н5.
Це МО № 15, для якої Е = -17.377
еВ.
Дипольні
моменти пероксикислот
Теоретична частина
Дипольні моменти використовуються
для того, щоб дати інформацію про структуру молекул, а також щоб перевірити
розраховані хвильові функції.
Згідно уявлень класичної фізики пара
електричних зарядів q,
рівних за абсолютною величиною, але протилежних за знаком і розміщених на
відстані r
один
від одного - це електричний диполь. Величина такого диполя визначається
електричним дипольним моментом - вектором, що характеризує зарядову асиметрію і
умовно спрямований від центра негативного до центра позитивного заряду.
Згідно Коулсону та Маллікену,
постійний електричний дипольний момент молекул речовини складається із
точкового та гібридизаційного. Причиною наявності точкового дипольного моменту
є зміщення центра заряду зв'язуючої пари електронів до атома з більшою
електронегативністю. Гібридизаційний дипольний момент молекулярних систем
обумовлений неспівпаданням центра тяжіння електронного розподілу атома з його
координатами.
У методі РМ3 дипольний момент
розраховується так:

Мета.
Освоєння методики квантовохімічного розрахунку дипольних моментів.
Завдання до
роботи.
1. Дипольні моменти та їх
компоненти для конформерів пероксикислот занести в таблицю.
. Провести аналіз впливу
конфігурації пероксикислот на величини дипольних моментів та їх компоненти.
Таблиця 7.
Дипольні моменти конформерів пероксикислот.
|
Параметр
|
(ОН)C6H4C(O)OOH
|
(CH3)(OH)C6H3C(O)OOH
|
|
Конформер А
|
Конформер Б
|
Конформер А
|
Конформер Б
|
|
 3.9724.2613.8264.557 3.9724.2613.8264.557
|
|
|
|
|
|
 2.706-3.0853.061-3.276 2.706-3.0853.061-3.276
|
|
|
|
|
|
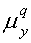 -1.4341.595-1.0751.948 -1.4341.595-1.0751.948
|
|
|
|
|
|
 -0.447-0.153-0.472-0.158 -0.447-0.153-0.472-0.158
|
|
|
|
|
|
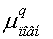 3.0953.4763.2783.815 3.0953.4763.2783.815
|
|
|
|
|
|
 0.671-0.8330.572-0.806 0.671-0.8330.572-0.806
|
|
|
|
|
|
 -0.5430.066-0.0950.068 -0.5430.066-0.0950.068
|
|
|
|
|
|
 -0.237-0.0600.210-0.048 -0.237-0.0600.210-0.048
|
|
|
|
|
|
 0.8950.8380.6170.810 0.8950.8380.6170.810
|
|
|
|
|
|
 3.377-3.9183.633-4.082 3.377-3.9183.633-4.082
|
|
|
|
|
|
 -1.9771.660-1.1702.016 -1.9771.660-1.1702.016
|
|
|
|
|
|
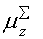 -0.685-0.213-0.262-0.206 -0.685-0.213-0.262-0.206
|
|
|
|
|
Висновки. Виконавши
цю роботу, мною були розраховані дипольні моменти та іх складові для
пероксикислот. Аналізуючи данні, наведені в таблиці 7, можна сказати, що
конформація пероксикислоти має значний вплив на значення дипольного моменту.
Для sp-конформацій
пероксикислот значення дипольних моментів меншими, ніж для ap-конформацій.
Енергія
хімічного зв’язку О-О пероксикислот
Теоретична частина.
Хімічний зв'язок - це така взаємодія
між атомами або молекулами, що призводить до створення з них стабільних чи
метастабільних хімічних сполук. Такими сполуками можуть бути як молекули, так і
конденсовані системи. Природа зв'язку (те, завдяки чому і як він виникає) є
об'єктом багаторазових обговорень і в науковій, і в навчальній літературі.
Існує декілька концепцій у теорії
хімічного зв'язку.
Маллікен запропонував наступну концепцію
природи хімічного зв'язку: хімічний зв'язок можна розглядати як суму
електростатичних та орбітальних взаємодій. Повну енергію хімічної частки можна
розрахувати як:


Також Маллікен запропонував
концепцію орбітального аналізу хімічних зв'язків - принцип відповідності
атомних орбіталей. Згідно із цим принципом, s- і
р-орбіталі бувають двох типів. Якщо АО однотипні, то їх комбінація зв'язуюча, а
якщо не однотипні - антизв'язуюча. Тип АО визначається знаком перед варіаційним
коефіцієнтом с у хвильовому рівнянні.
Мета: освоєння
методики розрахунку енергії хімічного зв’язку в наближенні орбітально-електростатичної
концепції Маллікена.
Завдання до
роботи.
.Енергію О-О зв’язку
та її складові компоненти конформерів пероксикислот занести в таблицю.
. Обгрунтувати вклад
електростатичних та орбітальних взаємодій в енергію зв’язку
-О-О-.
. Провести аналіз впливу
природи замісника і конформації пероксикислот на енергію -О-О- зв’язку.
Таблиця 8.
Енергетика О1-О2 зв’язку пероксикислот.
|
ЕАВ, еВ
|
(OH)C6H4C(O)OOH
|
(CH3)(OH)C6H3C(O)OOH
|
|
Конформер
|
Конформер
|
|
А
|
Б
|
А
|
Б
|
|
ЕJAB
|
-9.989
|
-10.067
|
-10.398
|
-10.063
|
|
ЕKAB
|
-4.434
|
-4.415
|
-4.487
|
-4.413
|
|
Еe,eAB
|
303.374
|
302.746
|
303.414
|
|
Еe,N
|
-594.110
|
-596.578
|
-597.117
|
-596.604
|
|
ЕN,NAB
|
297.716
|
298.526
|
299.878
|
298.511
|
|
ЕCAB
|
5.207
|
5.323
|
5.507
|
5.322
|
|
ЕАВ
|
-9.216
|
-9.159
|
-9.378
|
-9.155
|
Висновки.
Виконавши цю роботу, мною були розраховані енергії хімічного зв’язку
пероксикислот в наближенні орбітально-електростатичної концепції Маллікена. Як
ми можемо бачити з данних, котрі наведені в таблиці 8, конформація
пероксикислот суттєво не впливає на значення енергії та її складових.
Енергія
дисоціації пероксикислот
Теоретична частина.
Міцність хімічного зв'язку
визначається енергією, яку необхідно витратити, щоб розірвати цей зв'язок. Така
енергія зветься енергією дисоціації.
Розрив зв'язку може бути здійснений
шляхом гомолізу або гетеролізу.
Гомоліз - розрив зв'язку, який
полягає в розпаруванні електронів, що його утворюють, кожен з електронів далі
залишається на одному з відокремлених фрагментів.
Гетероліз - розрив зв'язку, що
супроводжується утворенням йонних частинок і відбувається таким чином, що пара
електронів, яка утворювала зв'язок, залишається на одному з фрагментів.
Мета.
Освоєння методики розрахунку енергії дисоціації хімічного зв’язку.
Завдання до роботи.
1. Розрахувати енергію більш
стабільного конформера пероксикислот.
2. Розрахувати енергію RС(О)ОО.
і RС(О)ОО-
в рівноважному стані.
3. Розрахувати енергію Н.
-радікала і Н+ - катіона.
Таблиця 9. Стандартні
ентальпії утворення пероксикислот та продуктів їх дисоціації
|
Об’єкт
|
∆Hf0,
ккал/моль
|
Об’єкт
|
∆Hf0,
ккал/моль
|
|
(ОН)C6H4C(O)OOH
|
-87.202
|
(CH3)(OH)C6H3C(O)OOH
|
-95.702
|
|
Н.
|
52.102
|
Н.
|
52.102
|
|
(ОН)C6H4C(O)OO·
|
-41.321
|
(CH3)(OH)C6H3C(O)OO·
|
-49.965
|
|
Н+
|
353.580
|
Н+
|
353.580
|
|
(ОН)C6H4C(O)OO-
|
-89.495
|
(CH3)(OH)C6H3C(O)OO-
|
-109.512
|

Для (ОН)C6H4C(O)OOH:
D1
= (353.580
+ (-89.495))
- (-87.202)
= 351.287 ккал/моль
Для (CH3)(OH)C6H3C(O)OOH:
D2
= (353.580
+ (-109.512))
- (-95.702) = 339.77 ккал/моль

Для (ОН)C6H4C(O)OOH:
D3
= (52.102 + (- 41.321)) - (-87.202) = 97.983
ккал/моль
Для (CH3)(OH)C6H3C(O)OOH:
D4
= (52.102 + (-49.965)) - (-95.702) = 97.839
ккал/моль
Висновки. Виконавши
цю роботу мною були розраховані енергії дисоціацій пероксикислот. Дивлячись на
отримані значення енергій можна стверджувати, що розрив зв'язку, здійснений
шляхом гомолізу, є енергетично вигіднішим, ніж шляхом гетеролізу. Тому, скоріше
за все, пероксикислоти будуть дисоціювати з утворенням радикалів.
Потенціали
іонізації пероксикислот
Теоретична частина.
Потенціал іонізації - енергія, яку
необхідно витратити, щоб іонізувати хімічну частинку шляхом відриву одного
електрона.
Внаслідок іонізації можлива зміна
геометрії та електронної структури частинок, що виникають. Тому розрізняють
адіабатичну та вертикальну іонізацію.
Адіабатичний потенціал іонізації -
різниця між енергіями іонізованої та неіонізованої форми хімічної частинки за
умови, що обидві частинки знаходяться у рівноважному стані.
Вертикальний потенціал іонізації -
енергія, яку потрібно затратити, щоб відняти від хімічної частинки електрон за
умови збереження нею початкової конфігурації.
Потенціал іонізації (за теоремою
Купманса) - це енергія НЗМО, взята з протилежним знаком.
Мета.
Освоєння методики розрахунку потенціалів іонізації хімічних сполук.
Завдання до роботи.
. Розрахувати
вертикальний потенціал іонізації пероксикислот.
2. Розрахувати
адіабатичний потенціал іонізації пероксикислот.
3. Визначити
потенціал іонізації за теоремою Купманса для пероксикислот.
Таблиця 10.
Повна енергія, стандартна теплота утворення і енергія ВЗМО пероксикислот та
відповідних їм катіон - радикалів
|
Об’єкти
|
Еповна, еВ
|
∆fH0,
ккал/моль
|
ЕНЗМО, еВ
|
|
(ОН)C6H4C(O)OOH
|
рівноважна конфігурація
|
-2093.960
|
-87.202
|
-9.577
|
|
(CH3)(OH)C6H3C(O)OOH
|
|
-2243.635
|
-95.702
|
-9.433
|
|
(OH)C6H4C(O)OOH+●
|
отримано шляхом вертикальної іонізації ARn
|
-2083.953
|
143.572
|
|
|
(CH3)(OH)C6H3C(O)OOH+●
|
|
-2231.566
|
182.600
|
|
|
(ОН)C6H4C(O)OOH+●
|
отримано шляхом адіабатичної іонізації ARn
|
-2084.886
|
122.063
|
|
|
(CH3)(OH)C6H3C(O)OOH+●
|
|
-2234.722
|
109.814
|
|
ІРад = 
ІРверт = 
Для (CH3)(OH)C6H3C(O)OOH:
ІРад = 109.814 - (-95.702) =
205.516 ккал/моль (8.913 еВ)
ІРверт = 182.600 - (-95.702) =
278.302 ккал/моль (12.069 еВ)
ІР(за теоремою Купманса) =
9.433 еВ
Для (OH)C6H4C(O)OOH:
ІРад = 122.063 - (-87.202) =
209.265 ккал/моль (9.074 еВ)
ІРверт = 143.572 -
(-87.202) =
230.774 ккал/моль (10.007 еВ)
ІР(за теоремою Купманса) = 9.577 еВ
Висновки. Виконавши
цю роботу, мною були розраховані потенціали іонізацій пероксикислот. Як
бачимо, потенціали іонізацій пероксикислот дуже близькі за значенням
один до одного. Крім того, значення ІР, розраховані за теоремою Купманса, також
є дуже близькими до значень ІР, розрахованих за допомогою повних енергій.
Енергія
електронної спорідненості хімічних сполук
Теоретична частина.
Електронна спорідненість - енергія,
що виділяється уразі приєднання одного електрона до хімічної частинки.
Згідно із принципом Франка-Кондона,
іонізація частинок перебігає без зміни їх геометрії. Цей принцип узгоджується з
вертикальним шляхом іонізації та виконується для сполук, що мають неподілену
пару електронів.
Адіабатична електронна спорідненість
- різниця між енергіями йонізованої та неіонізованої форми хімічної частинки за
умов, коли обидві частинки знаходяться в рівноважному стані
Вертикальна електронна спорідненість
- енергія, яка виділяється при приєднанні електрона хімічною частинкою за умови
збереження нею початкової конфігурації.
Електронна спорідненість (за
теоремою Купманса) - енергія НВМО, взята з протилежним знаком.
Мета.
Освоєння методики квантовохімічного розрахунку енергії електронної
спорідненості молекулярних систем.
Завдання до роботи.
1. Розрахувати вертикальну
електронну спорідненість пероксикислот.
. Визначити за теоремою
Купманса електронну спорідненість більш стабільних конформерів пероксикислот.
Таблиця 11. Стандартна
ентальпія утворення і енергія НВМО пероксикислот та їх відповідних
аніон-радикалів
|
Об’єкти
|
Інформація
|
∆fH0,
ккал/моль
|
ЕНЗМО, еВ
|
|
(OH)C6H4C(O)OOH
|
Рівноважна конфігурація
|
-87.202
|
-0.379
|
|
(ОН)C6H4C(O)OOH●
|
Вертикальна іонізація
|
-93.009
|
|
|
(CH3)(OH)C6H3C(O)OOH
|
Рівноважна конфігурація
|
-95.702
|
-0.633
|
|
(CH3)(OH)C6H3C(O)OOH-●
|
Вертикальна іонізація
|
-99.935
|
|
ЕАверт = 
Для (OH)C6H4C(O)OOH:
ЕАверт = -93.009 -
(-87.202) =
-5.807 ккал/моль
Для (CH3)(OH)C6H3C(O)OOH:
ЕАверт = -99.935 - (-95.702) =
- 4.233 ккал/моль
Висновки. Виконавши
цю роботу, мною були розраховані енергії електронної спорідненості
пероксикислот. ЕА пероксикислот є дуже близькими між собою за значеннями. ЕА
(за теоремою Купманса) знайти неможливо, бо енергія НЗМО < 0.
Заключення
Виконавши цю роботу, навчився
використовувати методи квантовохімічного молекулярного моделювання як засіб
отримання інформації про будову і властивості хімічних сполук. Однак, має місце
і той факт, що найбільш ефективне використання програм квантовохімічних
розрахунків можливе тільки у тому випадку, коли дослідник хоча б у загальних
рисах уявляє, які операції вони виконують, і як інтерпретувати отримані
результати.
При розрахунках та обчисленні
результатів набув висновку, що конформація даної пероксикислоти впливає на
параметри молекулярної геометрії та електронної структури пероксикислот, але
цей вплив не завжди є суттєвим.
Також на деякі параметри
молекулярної геометрії пероксикислот впливає і наявність замісників у молекулі.
Література
1.Практичний
курс комп’ютерної структурної хімії. Навчальний посібник / М.А. Туровський,
О.М. Туровська / Донецьк: ДонНУ, 2004. 131 с.
2.Минкин
В.И., Симкин Б.Я., Миняев Р.М. Теория строения
молекул (электронные оболочки): Учеб. Пособие для ун-тов. - М.: Высш. Школа,
1979. - 407с.